
New Tools for Detecting MultirefPredict Character in Transition Metal Chemical Space
Introduction
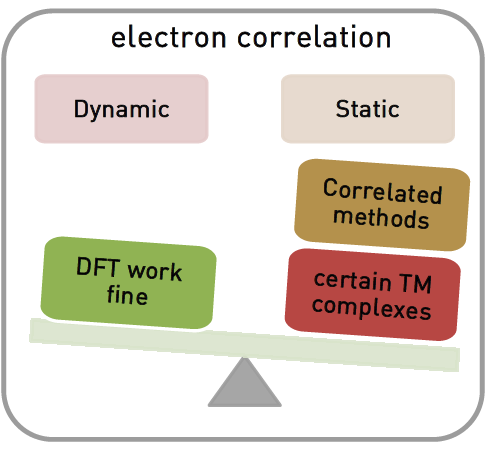
Open-shell transition metal complexes is essential for materials design and catalysis, but notoriously difficult to study accurately with electronic structure (ES) methods.
Computationally predicted molecular properties can be strongly affected the choice of ES methods. Density functional theory (DFT) results can vary significantly with the choice of functionals and parameters.
Impacts of method selection
For example, the spin-state energetic is defined as
\[\Delta E_{\textrm{H-L}}= E_{\textrm{HS}} - E_{\textrm{LS}}\]
First principle calculation
The DFT predicted spin-splitting energy shifts significantly with DFT parameters.
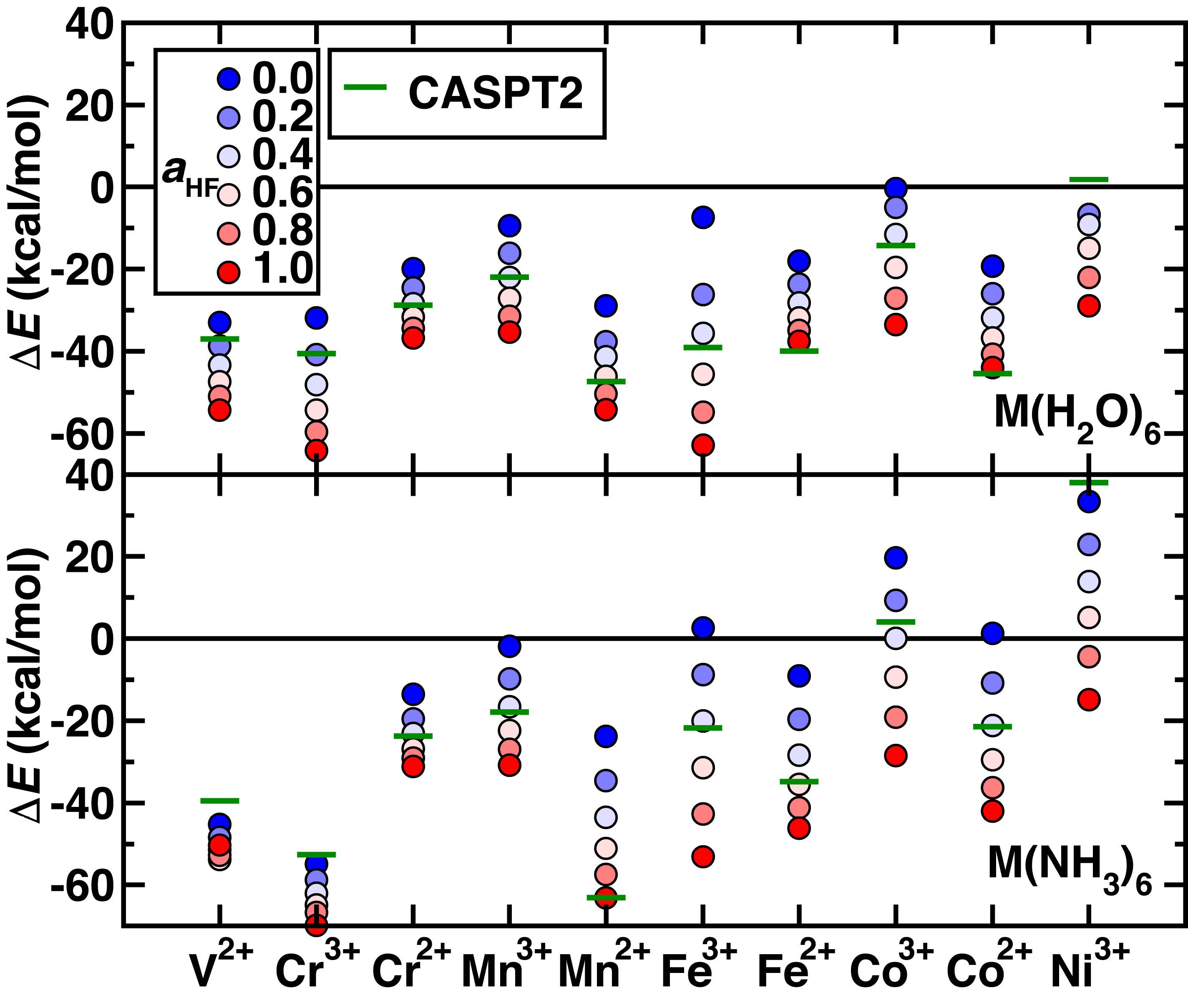 Figure 1: DFT results shift with parameters. F Liu, et al. Front. Chem. (2019))
Figure 1: DFT results shift with parameters. F Liu, et al. Front. Chem. (2019))
Machine learning
Performance of machine learning models trained on DFT calculated datasets also shift with parameters.
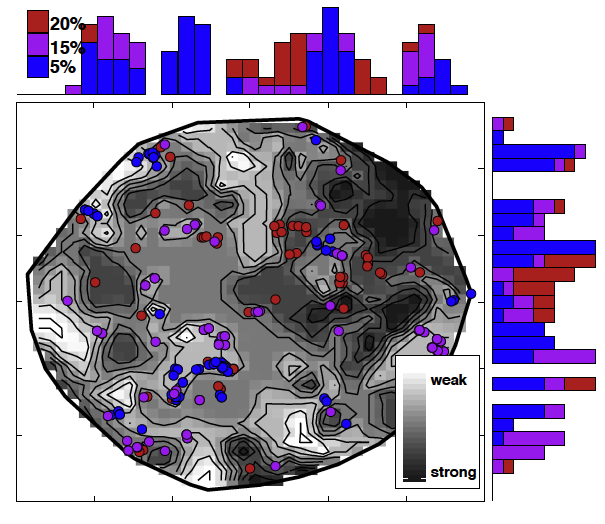 Figure 2: Machine learning predicted spin-crossover compound leads also shift with DFT parameters. J. P. Janet, F Liu et al., Inorg. Chem. (2019)
Figure 2: Machine learning predicted spin-crossover compound leads also shift with DFT parameters. J. P. Janet, F Liu et al., Inorg. Chem. (2019)
Objectives
A crucial step to obtain accurate results is to choose between DFT and correlated wavefunction based based methods.
Multireference character
Molecules with strong static correlation, i.e., multireference (MR) character, need to be calculated with correlated methods.
Decision Engineering
For high throughput computational design and discovery, an automated decision engine for method selection is needed.
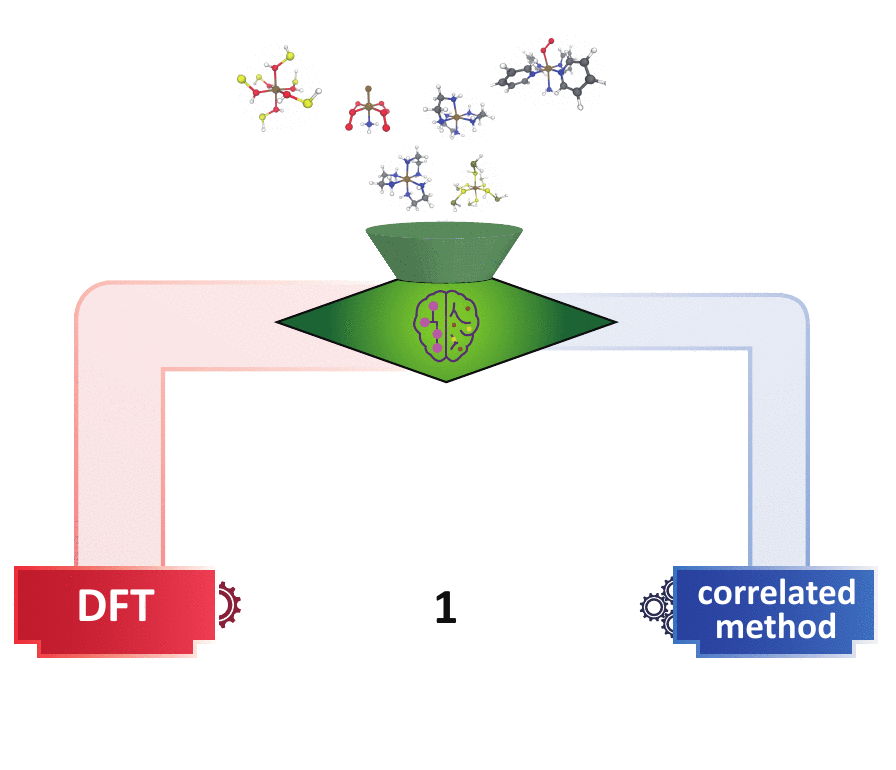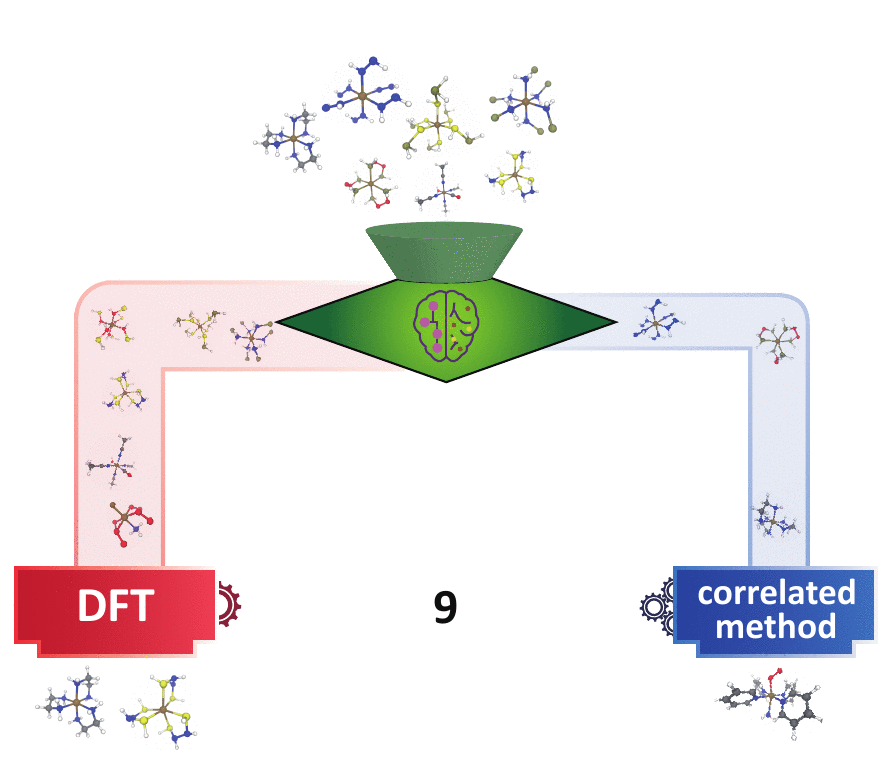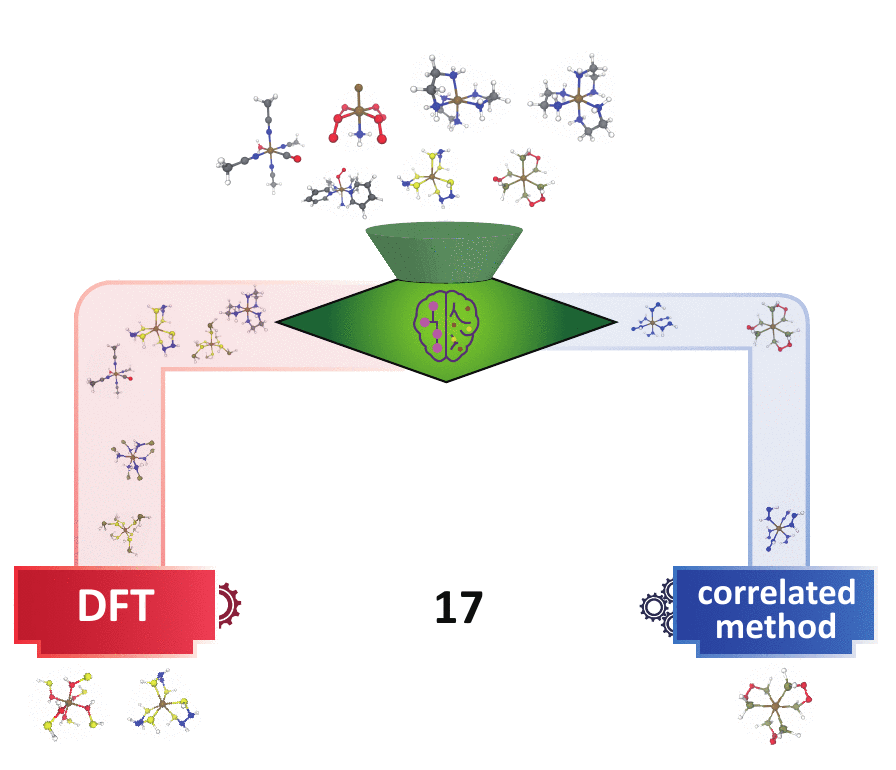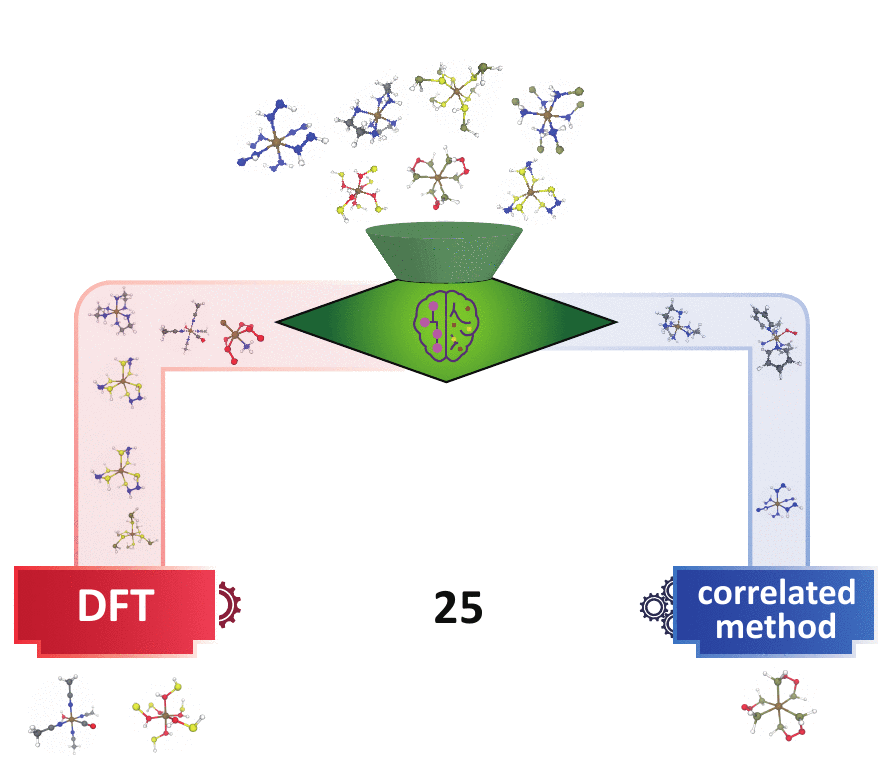
Methods
MR diagnostics: measurement of MR characters, essential for method selection
Energy based diagnostics
- BE
- bonding energy.
- TAE
- total atomization energy
-
\(B_1\) diagnostics
\[B_{1} = \frac{\text{BE}_{\text{BLYP}}-\text{BE}_{\text{B1LYP/BLYP}}}{n_{\text{bonds}}}\] -
\({A_{25\% }}[\rm{PBE}]\) diagnostics
\[{A_{25\% }}[\rm{PBE}] = 4\times \frac{\rm{1} - \rm{TAE[PBE0]}}{\rm{TAE[PBE]}}\] -
\(\% \rm{TAE}\) diagnostics
\[\% \rm{TAE} = 100 \times \frac{\rm{TAE}_{\rm{e}}\rm{[CCSD(T)]} - \rm{TAE}_{\rm{e}} \rm{[CCSD]}}{\rm{TAE}_{\rm{e}} \rm{[CCSD(T)}}\]
CASSCF based diagnostics
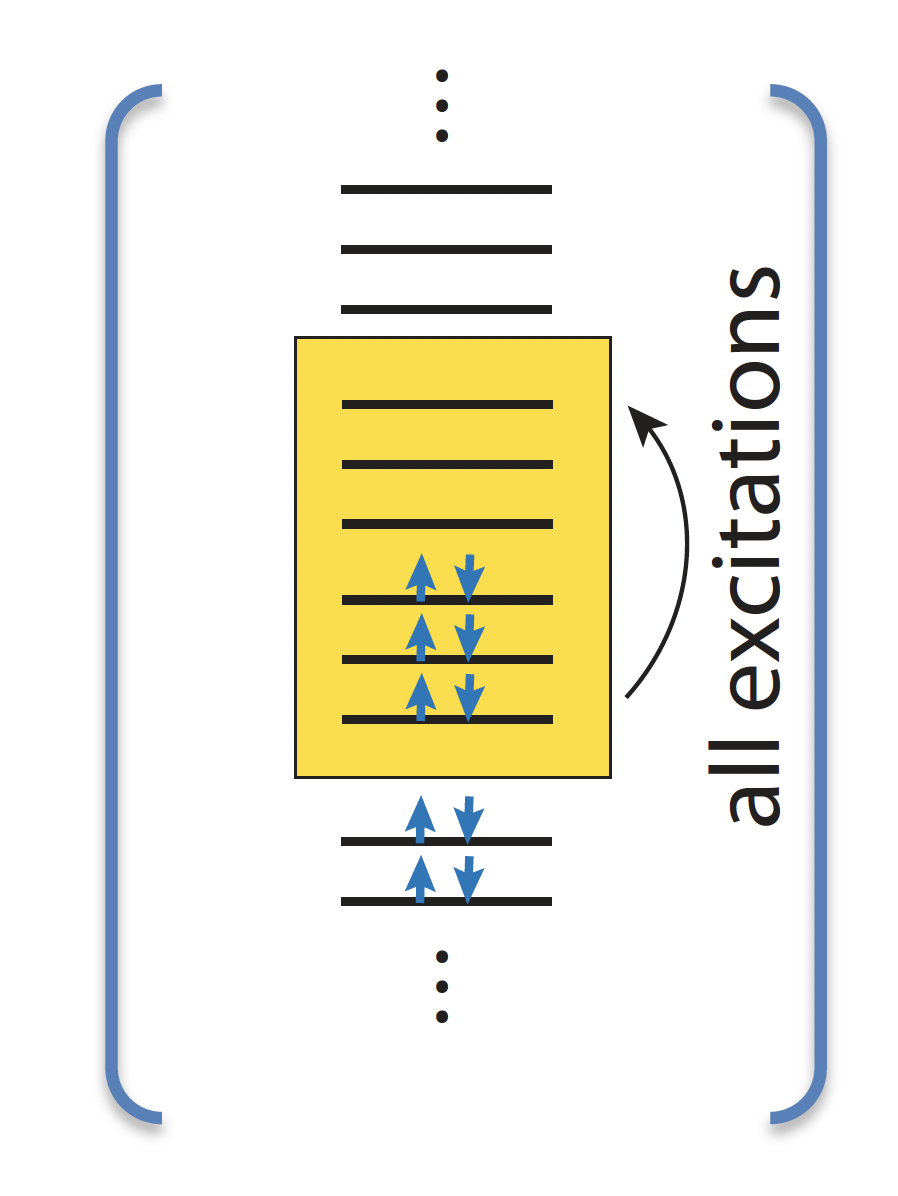
\(C_I\): CI coefficients
\(C_0^2\) diagnostics: square of the CI coefficient of the leading determinant
Coupled-cluster (CC) based diagnostics
\(\Psi _{\rm{CC}} \rangle = {e^{\hat T}}\left| {\Phi _I} \right\rangle ,\;{\kern 1pt} \hat T = {\hat T_1} + {\hat T_2} + {\hat T_3} + ...\)
-
\(T_1\) diagnostics
\[T_1 = \frac{\sqrt {\overrightarrow {t_1} \cdot \overrightarrow {t_1} } }{\sqrt {N_{\rm{corr}}} }\] -
\(D_1\) diagnostics
\[D_1\rm{(CCSD) } = \left\| \bf{T} \right\|_{2} , {\bf{T}}_{ia} = t_i^a\] -
\(D_2\) diagnostics
\[D_2 \rm{(CCSD) } = \rm{max} \{ D_2^o,D_2^v\} , \bf{T}_{jab,i}^o = \bf{T}_{ijb,a}^v = t_{ij}^{ab}\] \[D_2^o = \rm{max} \left\| {\bf{T}}^o{\bf{u}} \right\|_2, D_2^v = \rm{max} \left\| {\bf{T}}^v{\bf{v}} \right\|_2\]
Fractional occupation number (FON) based diagnostics
-
\(I_{\rm{ND}}\) diagnostics
\[I_{\rm{ND}} = \frac{1}{2}\sum\limits_{\sigma ,i} {n_i^\sigma } (1 - n_i^\sigma )\] \[I_{\rm{D}} = \frac{1}{4}\sum\limits_{\sigma ,i} {[n_i^\sigma (1 - n_i^\sigma )]^{1/2}} - \frac{1}{2}\sum\limits_{\sigma ,i} {n_\sigma ^i} (1 - n_\sigma ^i)\] -
\(r_{\rm{ND}}\) diagnostics
\[r_{\rm{ND}} = \frac{I_{\rm{ND}}}{I_{\rm{D}} + I_{\rm{ND}}}\] -
FOD (fractional occupation density) diagnostics
\[\rho _{\rm{FOD}} = \sum\limits_{\sigma ,i} {(\delta _1 - \delta _2 n_i^\sigma )|\phi _i^\sigma ({\bf{r}})|^2}\] \[N_{\rm{FOD}} = \int {\rho _{\rm{FOD}}({\bf{r}}){\rm{d}}{\bf{r}}}\]
QCEngine Interface for TeraChem
To utilize the advantages of different quantum chemistry (QC) packages, MultirefPredict conducts QC calculations through QCEngine, an open-source executor and IO standardizer. We implemented the interface for the GPU based QC package, TeraChem, in QCEngine to exploit the massive parallel power of GPUs.
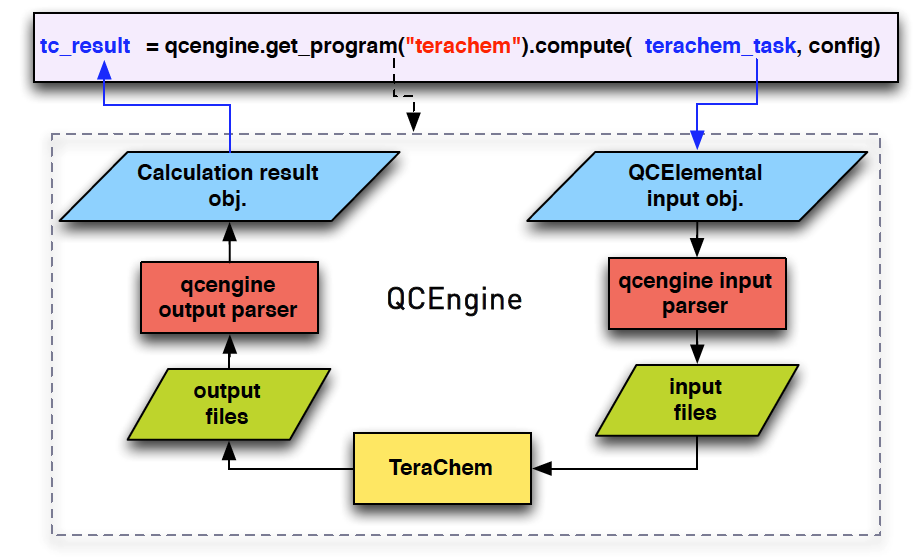
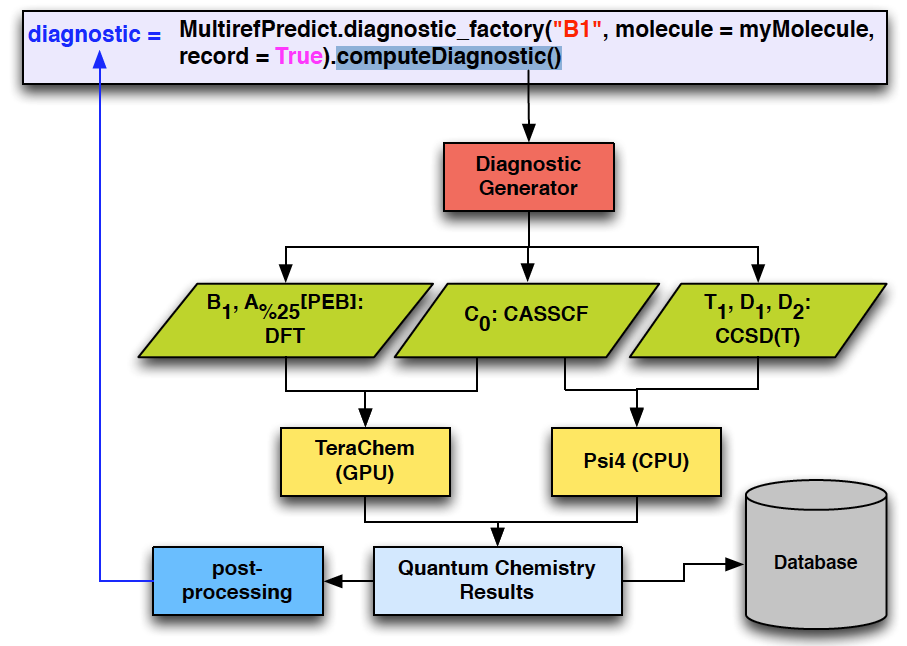
MultirefPredict
MultirefPredict is an open source python module we developed to automate the generation of commonly used multi-reference diagnostics.
- Source code available on Github
- Online documentation available
- MolSSI best practices for programming are enforced in the development: version control, testing and code coverage, continuous integration, code style, documentation (Github badges shown below).

- User-friendly interface. Automatically handling I/O for QC packages.
- Backend QC packages (TeraChem, Psi4, etc.) can be chosen by default based on availability of the software/hardware and the type of required calculation, or from user’s input.
Example
Example python script to calculate B1 diagnostics through MultirefPredict:
1
2
3
4
5
6
7
8
9
10
import MultirefPredict
import qcelemental
mol = qcelemental.models.Molecule.from_data("""
O 0.000000000000 0.000000000000 -0.068516245955
H 0.000000000000 -0.790689888800 0.543701278274
H 0.000000000000 0.790689888800 0.543701278274
""")
b1 = MultirefPredict.diagnostic_factory("B1",molecule=mol, molname="water", record=False).computeDiagnostic()
It takes only one command to define the molecule to calculate, and another line to tell MultirefPredict what diagnostics to calculate. No need to know the quantum chemistry packages to use, or the format of their input/output files.
Conclusions
- Implemented QCEngine interface for the GPU based quantum chemistry package TeraChem to enable easy control of TeraChem through python
- Developed automated module, MultirefPredict, to calculate MR diagnostics.
- Enabled automated workflow to generate transition metal complexes structure and estimate their multireference character.
References
- F. Liu, et al. DOI: 10.3389/fchem.2019.00219
- J. P. Janet, F. Liu, et al. DOI: 10.1021/acs.inorgchem.9b00109
- molSimplify: https://molSimplify.mit.edu
- C. Duan, F. Liu, et al. DOI: 10.1021/acs.jctc.0c00358
- E. Ramos-Cordoba, E. Matito, DOI: 10.1021/acs.jctc.7b00293
Acknowledgements
Software Fellow Fang Liu was supported by a fellowship from The Molecular Sciences Software Institute under NSF grant OAC-1547580”