
Tinker GPU: GPU-Accelerated MD Simulation Software for Advanced Force Fields and Enhanced Sampling
Introduction
Advanced potential energy surfaces and models are being applied to an ever-expanding array of problems in structural biology, drug design and characterization of novel materials. These new potentials incorporate explicit polarization effects and are calibrated very closely against energy decompositions derived from electronic structure calculations. The Tinker molecular modeling packages constitute one of the leading code families implementing the AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications) force field and many other advanced potential energy models.
As a new member of the Tinker suite, tinker.gpu
(get it on GitHub) will focus on
accelerating the advanced force fields and enhanced sampling methods with novel
parallel algorithms on the state-of-the-art GPU devices.
Advanced Molecular Modeling
The polarizable force field AMOEBA has been implemented in tinker.gpu. Our
next-generation force field HIPPO (Hydrogen-like Polarizable Potential)
developed in the Ponder lab, and other advanced force fields developed by our
collaborators are being added. In addition to the new energy terms,
we are in close collaboration with Prof. Wei Yang from Florida State University
to add his family of powerful orthogonal space sampling methods for biomolecular
applications.
Multipole Electrostatics
The pairwise multipole electrostatics (\(U_{ij}\)) includes up to quadrupole-quadrupole interaction,
\[U_{ij} = M_i T_{ij} M_j,\]where \(M\) is the multipole in Cartesian coordinate
\[M = [c, d_x, d_y, d_z, q_{xx}, q_{xy}, \cdots, q_{zz}],\]and
\[T_{ij} = \begin{pmatrix} 1 & \nabla_j & \nabla_j^2 \\ \nabla_i & \nabla_i \nabla_j & \nabla_i \nabla_j^2 \\ \nabla_i^2 & \nabla_i^2\nabla_j & \nabla_i^2\nabla_j^2 \end{pmatrix}\frac{1}{r_{ij}}.\]Polarization
If the system has \(N\) atoms, the induced dipoles \(\mu\) are first obtained by solving a \(3N \times 3N\) linear equation \(\mu = T_p^{-1} {\bf E}\), where \({\bf E}\) is the electrostatic field due to the permanent multipoles, and \(T_p\) is the matrix analogous to multipole interaction with Thole damping. [1] The polarization energy is computed by
\[U_p = -\frac{1}{2}\mu\cdot{\bf E}.\]Charge Transfer
The charge transfer term was added to HIPPO force field. It has an empirical pairwise form of
\[U_{ct} = -A_i\exp(-\eta_j r) -A_i\exp(-\eta_i r),\]where \(A\) is the magnitude coefficient and \(\eta\) is the exponential decay factor.
Dispersion and Repulsion
The dispersion and repulsion terms are also newly supported energy terms introduced by HIPPO. [2] Dispersion (and Dispersion PME) are implemented as the classical dispersion interaction, plus extra damping function to account for the quantum effects. The form of damping function adopted in HIPPO is
\[f(r) = 1 - (1+\frac{1}{2}\alpha r)\exp(-\alpha r).\]We use the pseudo-orbital model with a form of
\[\phi = \sqrt{\frac{Q\alpha^3}{8\pi}} \exp(-\alpha r/2)\]to account for the repulsion energy potential.
Code Structure
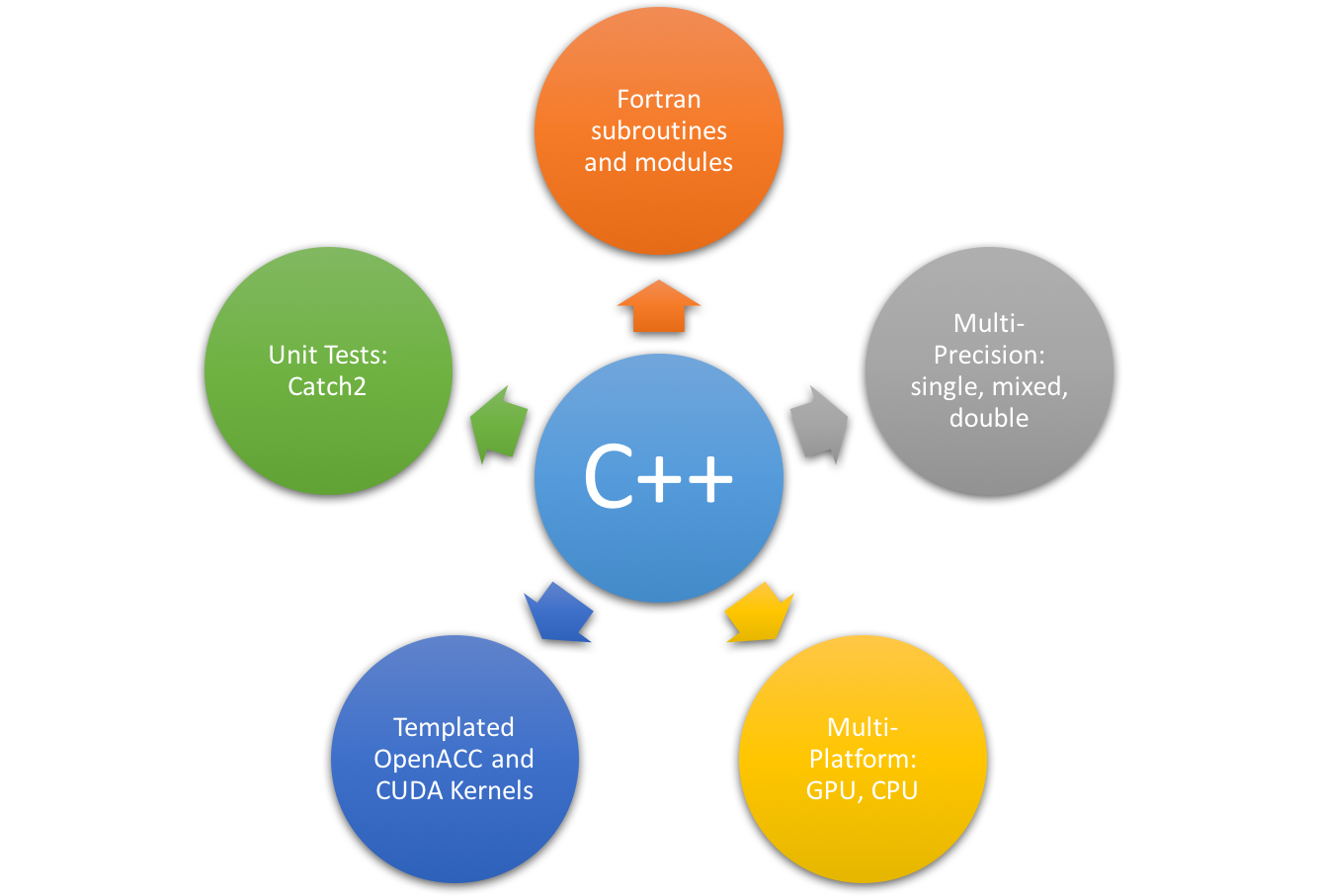
Even though tinker.gpu is written in modern templated C++, we will not
immediately give up our Fortran code base. As a matter of fact, this
program still depends on the pre-built Fortran Tinker subroutines and modules
to manage the program I/O and global variables. The C++ is the glue layer to
communicate with Fortran library, to launch OpenACC/CUDA kernels at runtime,
and to perform unit tests.
Fortran Subroutines and Modules
The interoperability of Fortran with C is of vital importance. Fortran subroutines with no character string parameters can often be used directly in C/C++ code with the assistance of following macro
// subroutine "getxyz" becomes "getxyz_" in C
// "call getxyz" becomes "TINKER_RT(getxyz)();"
#define TINKER_RT(getxyz) getxyz##_
whereas for the global variables in the modules, different compiler vendors may
have different implementations. We used the following macro to access variable
var in module mod, for gfortran and Intel ifort, respectively
// if gfortran
#define TINKER_MOD(mod, var) __##mod##_MOD_##var
// if Intel ifort
#define TINKER_MOD(mod, var) mod##_mp_##var##_
A syntax parser that can scan the Fortran Tinker source code and generate the aforementioned bindings has been added to the source code.
The alternative solution would be exporting these symbols to C programs from
Fortran source code using iso_c_binding but requires extensive modification
in the Fortran code base.
Multi-precision and Multi-Platform
Our program can be configured (by CMake options) to compile with single precision, double precision, and mixed precision floating-point numbers, as well as to compile the production GPU build or reference CPU build.
Templated OpenACC and CUDA Kernels
Here are some of our principles in the GPU kernel developments that lead us to the multipole kernel shown below.
- Don’t repeat ourselves, especially for the energy/gradient/virial expressions.
- Don’t pay for the things we don’t need, e.g., skip energy evaluation in the simulation between saved MD frames.
- Put calculation on GPU as much as possible.
- Maximal code reuse in multipole electrostatics.
- Minimal communication and synchronization between CPU and GPU.
- Always implement an OpenACC kernel first, unless we believe there will be significant improvement by CUDA.
/// Pairwise quadrupole electrostatics.
/// @tparam Version Flag for using energy counts, potential energy,
/// virial tensor, and energy gradient in the pairwise
/// interaction.
/// @tparam ElecType Flag for doing real space PME or Non-EWALD pairwise
/// electrostatics.
template <class Version, class ElecType>
__device__
void pair_mpole(INPUT_PARAMETERS, RESULTS)
{
constexpr bool do_a = Version::a; // flag for number of interactions
constexpr bool do_e = Version::e; // flag for potential energy
constexpr bool do_v = Version::v; // flag for virial tensor
constexpr bool do_g = Version::g; // flag for energy gradient
// ...
}
Unit Tests and Continuous Integration
We are using Catch2 as the unit test framework and Travis CI as the continuous integration platform for the reference CPU build.
Conclusion
This program tinker.gpu, has proved feasible and effective to incorporate
recent software advances and enable community collaboration. It is a convenient
framework to implement novel advanced force fields of greater complexity with
lower engineering difficulty and maintain good performance.
Benchmark results are comparable to our previous work. [3]
References
- Polarizable Atomic Multipole Water Model for Molecular Mechanics Simulation. Ren et al. J. Phys. Chem. B, 107, 5933-5947 (2003)
- A Physically Grounded Damped Dispersion Model with Particle Mesh Ewald Summation. Rackers et al. J. Chem. Phys. 149 084115 (2018); Classical Pauli Repulsion: An Anisotropic, Atomic Multipole Model. Rackers et al. J. Chem. Phys. 150 084104 (2019)
- Tinker 8: Software Tools for Molecular Design. Rackers et al. J. Chem. Theory Comput., 14, 5273-5289 (2018)
Acknowledgements
Zhi Wang was supported by a fellowship from The Molecular Sciences Software Institute under NSF grant OAC-1547580.
