Efficient implementation of ADC for ionization and electron attachment in molecules
Introduction
- Accurate computations of ionization potential (IP) and electron attachment (EA) are important for predicting properties of molecules and materials, such as redox potentials, band gaps, and photoelectron spectra.
- Development of reliable and efficient theoretical methods for computing IP and EA of molecules and materials is one of the current challenges in modern quantum chemistry.
- To address this problem, we developed a new and efficient computer implementation of algebraic diagrammatic construction theory (ADC) for simulating IP/EA of molecules in the open-source quantum chemical software package PySCF.
Theory
- Spectroscopic studies probe a molecular system by subjecting it to an external perturbation such as electromagnetic field and then studying the response of the system to that perturbation.
- The mathematical function required for computing the response is called a propagator.
- The IP/EA-ADC approximations are derived from a perturbative expansion of the one-electron propagator, poles and residues of which provide information about ionization and electron-attachment energies and transition intensities.
Implementation
- The present ADC implementation in PySCF can be applied to both closed- and open-shell molecules starting with either restricted or unrestricted Hartree-Fock orbitals.
- The key step of my IP/EA-ADC implementation is a function that defines the form of the \(\boldsymbol{\sigma} = \textbf{MX}\) vector that is the product of the ADC effective Hamiltonian matrix (\(\textbf{M}\)) and a trial vector (\(\textbf{X}\)).
- Once the \(\boldsymbol{\sigma}\) vector is defined for a particular ADC approximation, conventional iterative diagonalization techniques are used to solve for several lowest IP or EA energies and the intensities of these transitions.
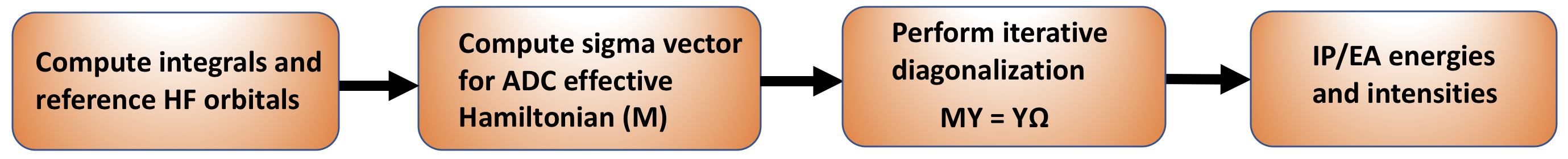
Figure 1: A schematic representation of the IP/EA-ADC implementation in PySCF.
Efficiency improvements to the previous IP/EA-ADC implementation
The pilot implementation of IP/EA-ADC was limited to systems with upto \(\sim\) 300 orbitals. As a part of my project supported by MolSSI, I made several improvements to increase the efficiency of my implementation in PySCF :
-
Development of a spin-restricted ADC (RADC) code for closed-shell molecules
-
An in-core algorithm with optimized memory and CPU efficiency
-
An out-of-core algorithm that reduces the memory requirements by storing two-electron integrals on disk (using \(\textit{ h5py}\) Python module capabilities) and computing them on-the-fly. This is done using a buffering algorithm that computes subsets of the two-electron integrals which fits in the available memory.
-
Use of Basic Linear Algebra Subroutines (BLAS) operations for expensive tensor contractions, which speeds up the calculations of the matrix-vector products (\(\boldsymbol{\sigma}\) = \(\textbf{MX}\))
-
A modified ADC code interface to allow for a direct calculation of excitation energies and properties
-
A modified preconditioner for Davidson’s iterative algorithm for better convergence of IP/EA energies
Results
-
The several improvements discussed above have led to significant computational savings both with respect to memory usage as well as computational wall times.
-
The new ADC implementation is more efficient than the highly optimized equation-of-motion coupled-cluster singles and doubles (EOM-CCSD) implementation in PySCF and has a similar accuracy.
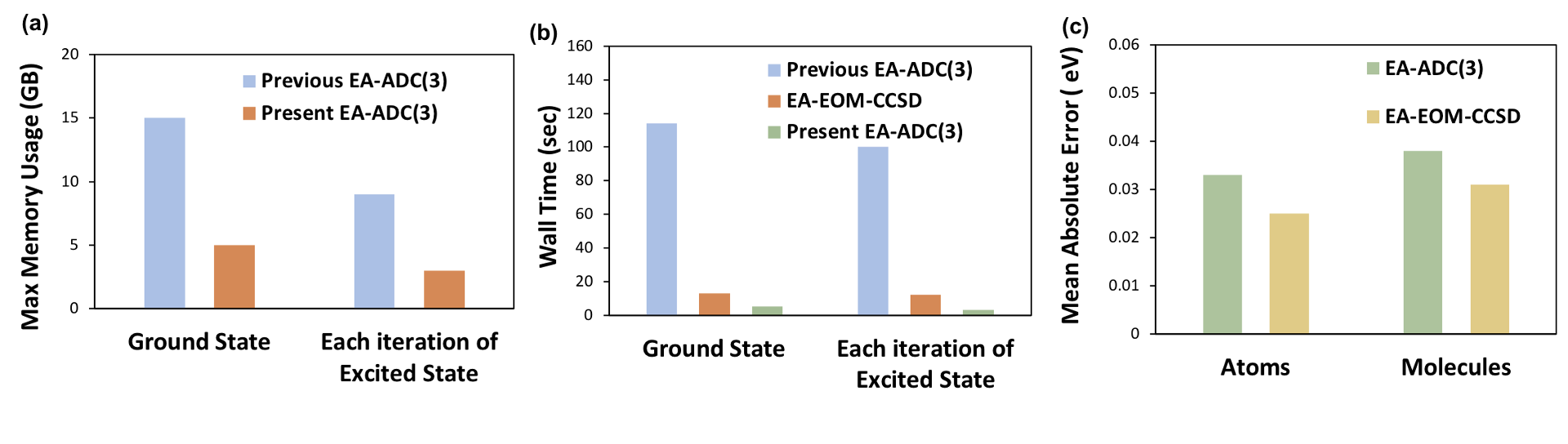 Figure 2: Comparison of
(a) maximum memory usage between the previous and present EA-ADC(3) implementations for H2CO (basis set: aug-cc-pVTZ),
(b) computational wall times between the previous and present EA-ADC(3) as well as EA-EOM-CCSD implementations for H2CO (basis set : aug-cc-pVTZ),
(c) mean absolute errors (eV) in EA’s for a set of closed-shell atoms and molecules simulated using EA-EOM-CCSD and EA-ADC(3) (basis set : aug-cc-pVQZ)
Figure 2: Comparison of
(a) maximum memory usage between the previous and present EA-ADC(3) implementations for H2CO (basis set: aug-cc-pVTZ),
(b) computational wall times between the previous and present EA-ADC(3) as well as EA-EOM-CCSD implementations for H2CO (basis set : aug-cc-pVTZ),
(c) mean absolute errors (eV) in EA’s for a set of closed-shell atoms and molecules simulated using EA-EOM-CCSD and EA-ADC(3) (basis set : aug-cc-pVQZ)
- The present ADC implementation allows simulation of larger systems (600 orbitals) compared to the previous implementation which was limited to 300 orbitals.
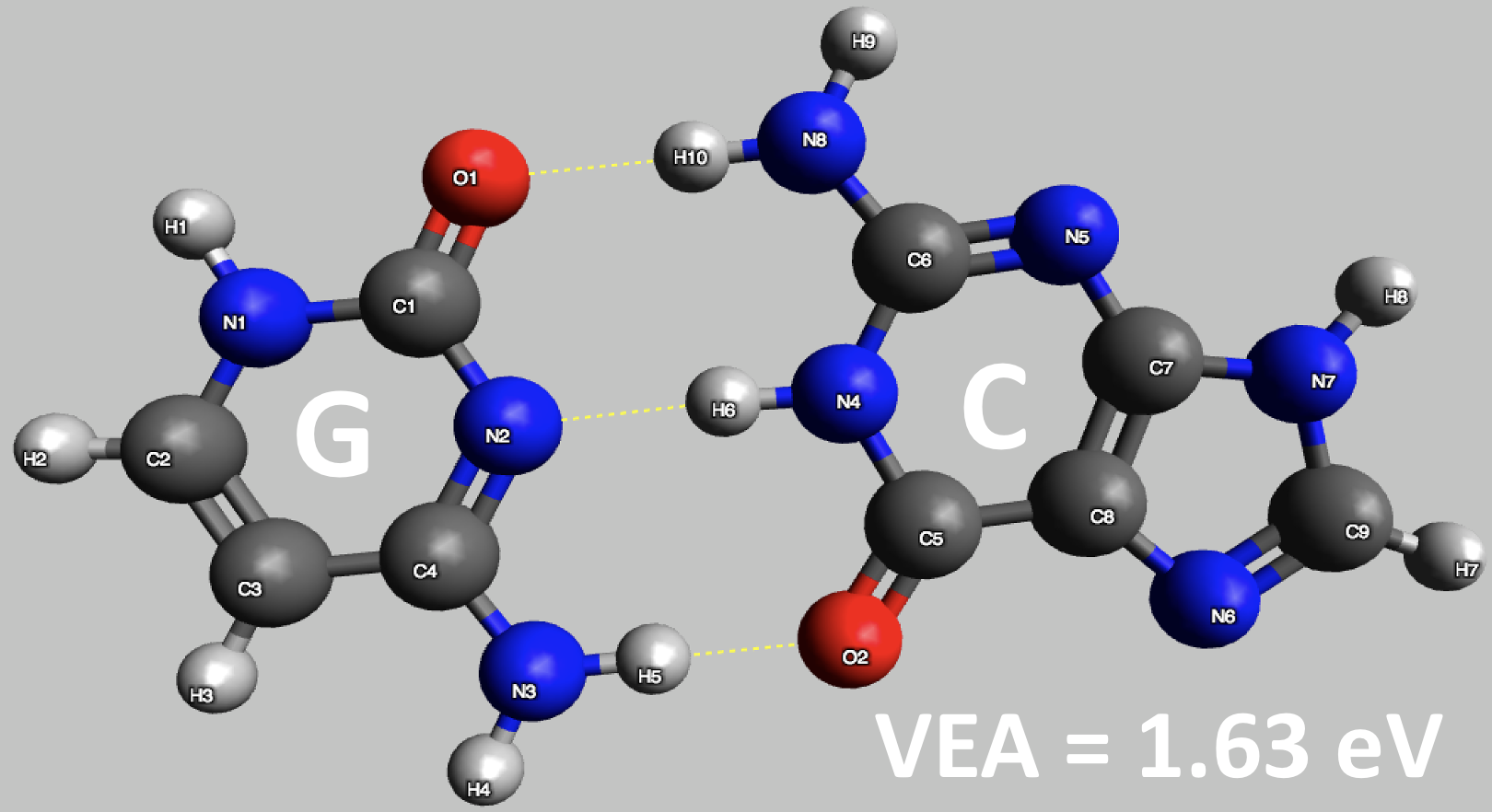
Figure 3: Preliminary results for vertical electron attachment energy of DNA nucleobase pair of guanine (G) and cytosine (C) computed using ADC(2) (basis set : aug-cc-pVDZ)
Conclusion
-
Implemented ADC as a new module in PySCF for accurate IP/EA simulation of molecules.
-
Made several efficiency modifications leading to significant reduction in memory requirements and computational wall times.
-
All the improvements enabled simulations of large molecules (600 orbitals), which paves the way for materials simulation using ADC in the future.
References
Acknowledgements
“Samragni Banerjee was supported by a fellowship from The Molecular Sciences Software Institute under NSF grant OAC-1547580”

![]()