
DESPASITO, Quickly Fit MD-Simulation Parameters using Equations of State
Introduction
First open-source application of its kind for Statistical Associating Fluid Theory (SAFT) equation of state (EOS).
Despite the power of SAFT coarse graining, its use has been limited by lack of availability of a freely accessible application.
![]() Figure 1: DESPASITO: Determining Equilibrium State and Parametrization: Application for SAFT, Intended for Thermodynamic Output
Figure 1: DESPASITO: Determining Equilibrium State and Parametrization: Application for SAFT, Intended for Thermodynamic Output
Project Goals:
- Design a thermodynamic platform for equations of state that allows natural extension
- Provide accessible parametrization routine that fits equation of state parameters that are useful for simulations
Audience:
- Historically complex equations of state are desired by the oil and gas industry
- Academically the SAFT EOS has been developed into a coarse-graining formalism [1-3] that provides chemical specificity to larger scale molecular dynamics simulations
Available Alternatives:
- Bottled SAFT [4] is a web application meant to remedy issue of parameterizing by applying corresponding states for homonuclear molecules, but represents a general correlation that relies on possessing critical properties.
- PC-SAFT calculations are available in the commercial package, Aspen
- The commercial package gSAFT from Process Systems Enterprise, PSE, is a more direct comparison supporting SAFT-γ-Mie
Statistical Associating Fluid Theory, SAFT
- Explicitly links the intermolecular potential with thermodynamic properties
- Parameters are fit to experimental vapor-liquid equilibria data and other macroscopic properties
- Various versions of SAFT can be used to obtain parameters for coarse-grained MD simulations
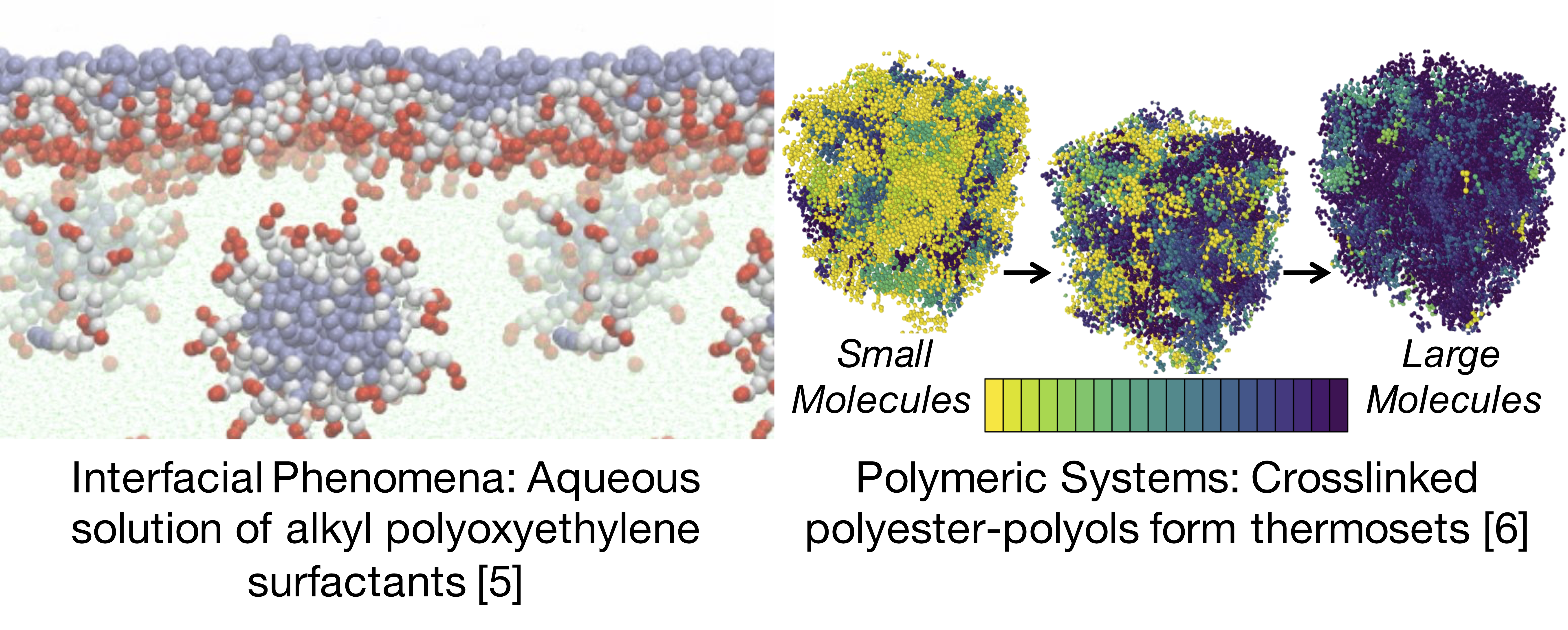 Figure 2: SAFT-γ-Mie has been successfully applied to various larger scale systems, DESPASITO makes contributing more accessible.
Figure 2: SAFT-γ-Mie has been successfully applied to various larger scale systems, DESPASITO makes contributing more accessible.
- As of now, a new contributor to this emerging method of coarse-graining would need to write their code from scratch
- Our application makes SAFT accessible to the general public
How it Works
Our modular and object oriented approach allows this package to function for an EOS other than SAFT, and expand the thermodynamic properties it handles.
- Want a different method of fitting? Add another function and our factory method will find it
- Want another objective function? Add a class using our template
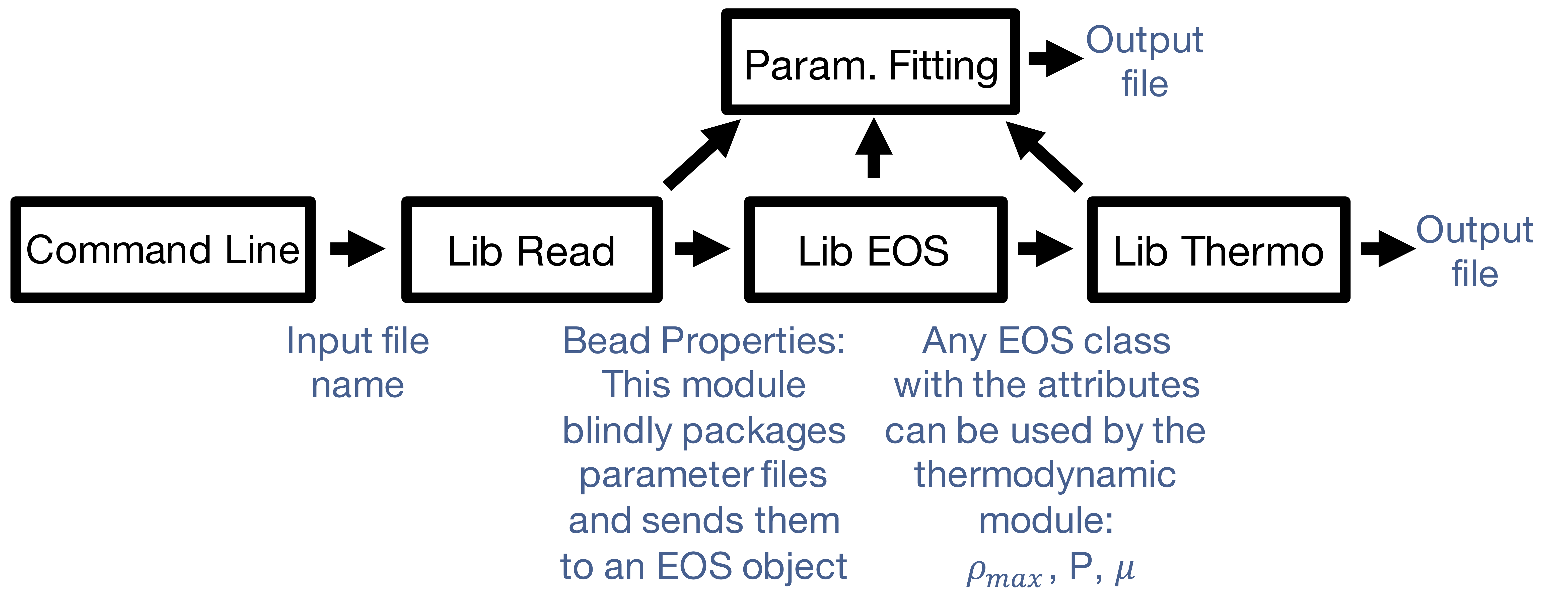 Figure 3: DESPASITO Program Flowchart
Figure 3: DESPASITO Program Flowchart
- Want a new calculation type? Add another function and our factory method will find it
- Want a new EOS? Use the class template for guaranteed compatibility
Example Calculations
For a united atom calculation of pentane and methane, a thermodynamic calculation is as easy as a 6 line file, input.json (given parameter files):
{
"beadconfig": [[["CH3", 2],["CH2", 3]], [["CH4", 1]]],
"EOSgroup": "SAFTgroup.json",
"EOScross": "SAFTcross.json",
"calculation_type" : "vapor_properties",
"Tlist" : [270.0],
"yilist": [[0.99, 0.01]]
}
Run it with:
$ python -m despasito -i input.json
DESPASITO can also be imported as a library so that a user can use the EOS object’s accessible attributes.
For example, we can break-down the Helmholtz energy components for hexane.
beads = ['CH3', 'CH2']
beads_per_molecule = np.array([[2., 8.]])
beadlibrary = {'CH3':
{'epsilon': 256.7662,'l_a': 6.0, 'l_r': 15.04982, 'sigma': 4.077257e-1, 'Sk': 0.5725512, 'Vks': 1, 'mass': 0.015035},
'CH2':
{'epsilon': 473.3893, 'l_a': 6.0, 'l_r': 19.87107, 'sigma': 4.880081e-1, 'Sk': 0.2293202, 'Vks': 1, 'mass': 0.014027}}
crosslibrary = {'CH3': {'CH2': {'epsilon': 350.770}}}
eos = despasito.equations_of_state.eos(eos="saft.gamma_mie", beads=beads , nui=beads_per_molecule , beadlibrary=beadlibrary , crosslibrary=crosslibrary )
rho, T, xi = 553.0, 700.0, np.array([1.0])
AHS = eos.saft_source.Ahard_sphere(rho, T, xi)
A1 = eos.saft_source.Afirst_order(rho, T, xi)
A2 = eos.saft_source.Asecond_order(rho, T, xi)
A3 = eos.saft_source.Athird_order(rho, T, xi)
Am1 = AHS+A1+A2+A3
Am2 = eos.Amonomer(rho, T, xi)
print('The Monomer Contribution for Helmholtz Energy: {},\n equals the sum of its components: {}'.format(Am2,Am1))
The Monomer Contribution for Helmholtz Energy: [-0.19323846],
equals the sum of its components: [-0.19323846]
These parameters were taken from Dufal et al. [3]
Currently Supported Features
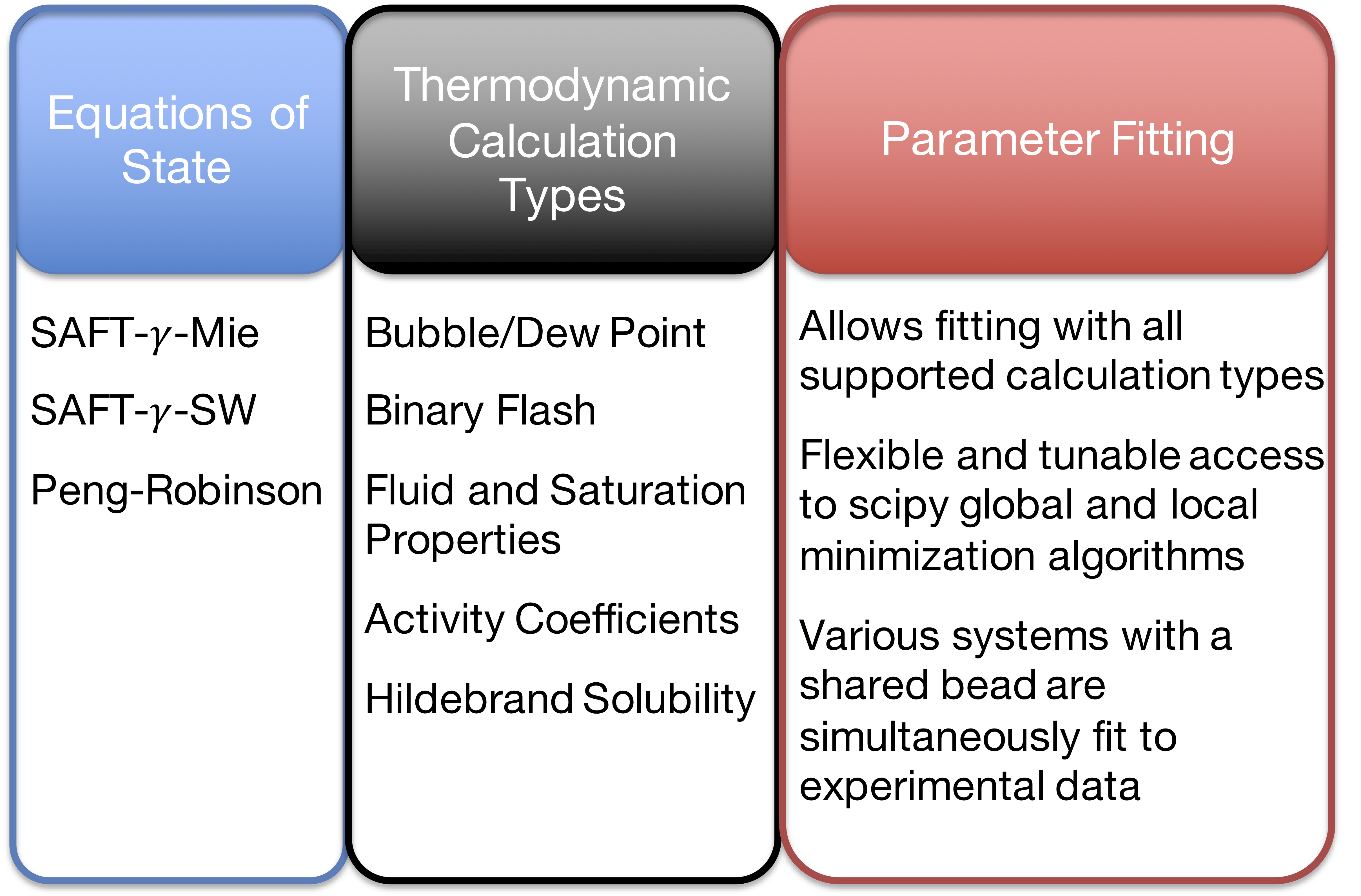
- Although our intention was to create a platform to fit parameters for the SAFT EOS, we are equipped to handle other equations of state that can’t be solved explicitly (e.g. Extended UNIQUAC).
- We added the Peng-Robinson EOS as an example of a non-SAFT EOS.
- Code Optimization: Implemented options to provide Numba, Cython, or Fortran modules.
- Shared-Memory Parallelization: Numerous thermodynamic or parametrization calculations can be run in parallel with multi cores for HPC resources.
What is Next?
- Add second derivative thermodynamic properties
- Add ability to implement complex parametrization constraints
- Upgrade thermodynamic algorithms for more robust multicomponent handling
- Provide interface for MoSDeF [7-8] for easy transfer from EOS to simulation
References
- Papaioannou, V.; Lafitte, T.; Adjiman, C. S.; Galindo, A.; Jackson, G. Simultaneous Prediction of Phase Behaviour and Second Derivative Properties with a Group Contribution Approach (SAFT-γ Mie). In Computer Aided Chemical Engineering; Elsevier, 2011; Vol. 29, pp 1593–1597.
- Papaioannou, V.; Lafitte, T.; Avendaño, C.; Adjiman, C. S.; Jackson, G.; Müller, E. A.; Galindo, A. Group Contribution Methodology Based on the Statistical Associating Fluid Theory for Heteronuclear Molecules Formed from Mie Segments. J. Chem. Phys. 2014, 140 (5), 054107.
- Dufal, S.; Papaioannou, V.; Sadeqzadeh, M.; Pogiatzis, T.; Chremos, A.; Adjiman, C. S.; Jackson, G.; Galindo, A. Prediction of Thermodynamic Properties and Phase Behavior of Fluids and Mixtures with the SAFT-γ Mie Group-Contribution Equation of State. J. Chem. Eng. Data 2014, 59 (10), 3272–3288.
- Ervik, Å.; Mejía, A.; Müller, E. A. Bottled SAFT: A Web App Providing SAFT-γ Mie Force Field Parameters for Thousands of Molecular Fluids. J. Chem. Inf. Model. 2016, 56 (9), 1609–1614.
- Müller EA; Jackson G. Force-Field Parameters from the SAFT-γ Equation of State for Use in Coarse-Grained Molecular Simulations. Annual Review of Chemical and Biomolecular Engineering. 2014, 5 (1), 405-427.
- Pervaje, A. K.; Tilly, J. C.; Detwiler, A. T.; Spontak, R. J.; Khan, S A.; Santiso, E. E. Molecular Simulations of Thermoset Polymers Implementing Theoretical Kinetics with Top-Down Coarse-Grained Models. Macromolecules. 2020, 53, 2310−2322.
- Klein C.; Sallai J.; Jones T.J.; Iacovella C.R.; McCabe C.; Cummings P.T. (2016) A Hierarchical, Component Based Approach to Screening Properties of Soft Matter. In: Snurr R., Adjiman C., Kofke D. (eds) Foundations of Molecular Modeling and Simulation. Molecular Modeling and Simulation (Applications and Perspectives). Springer, Singapore
- Klein C.; Summers, A. Z.; Thompson, M. W.; Gilmer, J. B.; McCabe, C.; Cummings, P. T.; Sallai, J.; Iacovella, C. R. Formalizing atom-typing and the dissemination of force fields with foyer. Computational Materials Science. 2019, 167, 215-227.
Acknowledgments
Jennifer Clark was supported by a fellowship from The Molecular Sciences Software Institute under NSF grant OAC-1547580
Jennifer Clark acknowledges North Carolina State University’s High Performance Computing (HPC) Services