
Using Machine Learning to Improve Coarse-Grained Simulations
Introduction
Machine learning is a powerful and flexible toolset that can be used alongside molecular dynamics simulations to improve accuracy and learn underlying distributions and characteristics of a system of interest. Here we demonstrate the use of a new software method, HTF,1 that connects the machine learning library TensorFlow with the molecular dynamics engine HOOMD-blue, with specific application to Coarse-Grained simulations.
Coarse-Grained simulations
A coarse-grained (CG) molecular dynamics (MD) simulation is a reduced dimensional approximation to a normal all-atom (AA) MD simulation.2-4 CG is a multiscale modeling technique which allows MD simulations at larger time-scales and length-scales.
 Figure 1: Illustration of different coarse-grained resolutions and how they are connected to different length-scale features.
Figure 1: Illustration of different coarse-grained resolutions and how they are connected to different length-scale features.
There are two steps that specify a coarse-grained (CG) system:
(i) Mapping operator: r → R
(ii) Potential energy: U(R)
HTF Software
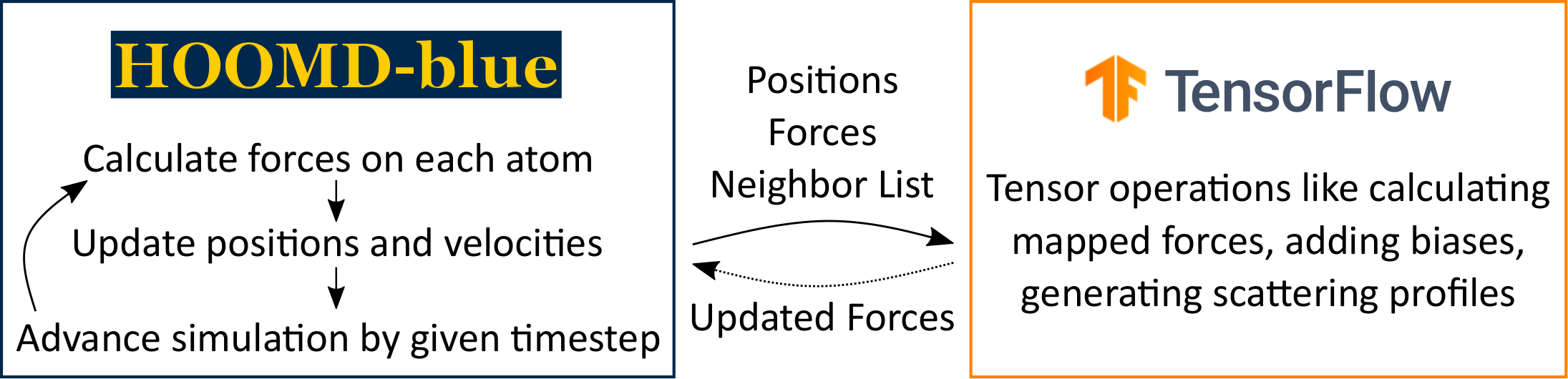
The HTF package gives TensorFlow5 access to the per-particle positions, neighbor lists, and forces generated by HOOMD-blue6 at each time step of a simulation. Hence, one can do learning on MD trajectories, propagate MD based on ML models, or both.
One of the challenges in CG simulations is the difficulty in accurately calculating CG potentials from AA simulation trajectories. Using HTF, one can process AA trajectories from popular simulation engines like GROMACS, learn the CG potential from this AA trajectory and run a CG simulation.
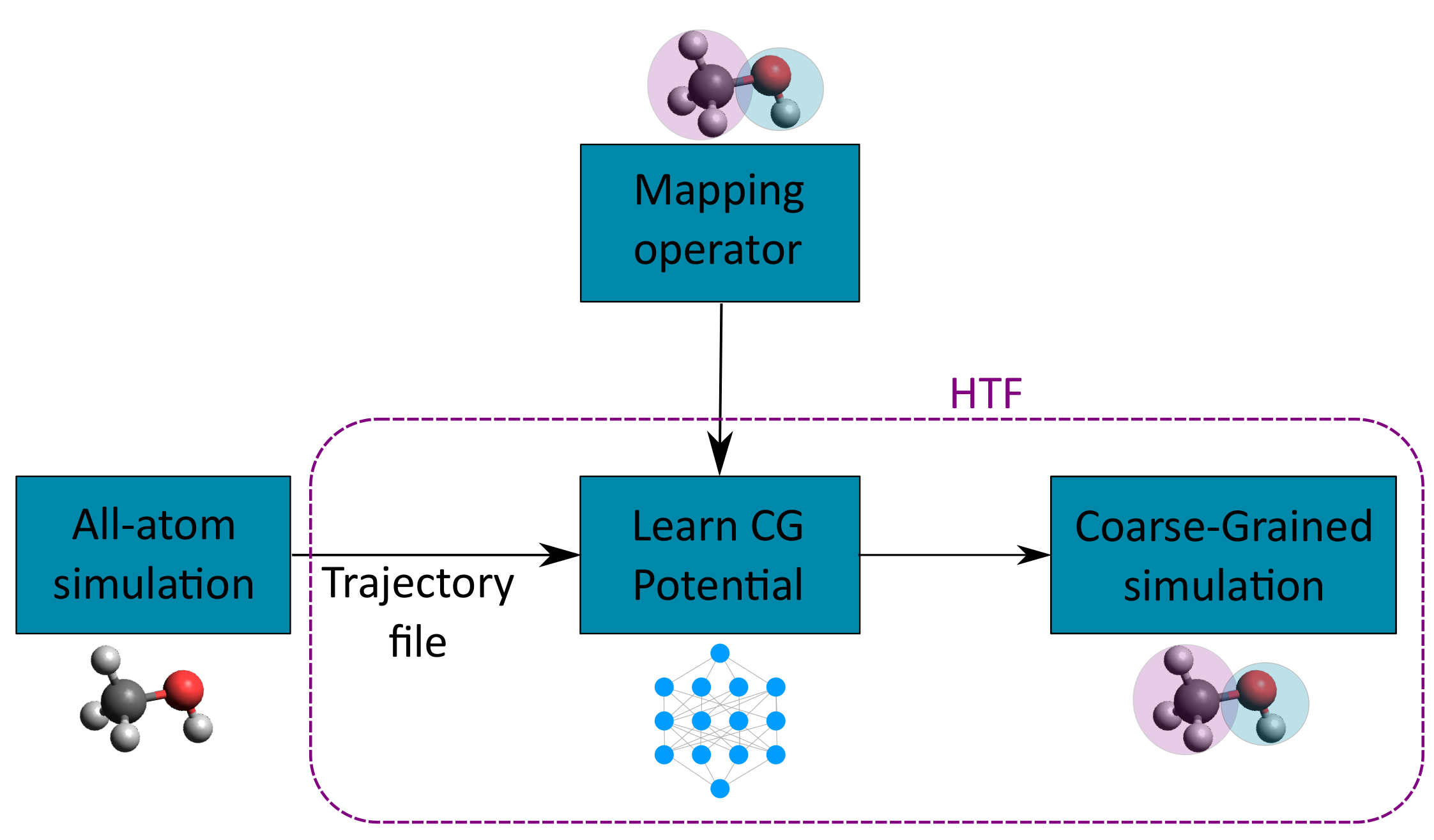
Using All-Atom Trajectories
The user needs to pass in a MDAnalysis7-8 Universe object to process the trajectory and perform any tensor operations on it which are specified in a TensorFlow graph. A complete example can be found in this tutorial.
Force Matching
To learn the CG potential we used Force Matching (FM). FMx is a bottom-up CG approach which aims to match the forces on CG particles as closely as possible to the cumulative forces on their constituent atoms in the reference AA simulations.
The reference mapped force is given by
\[\mathbf{F}^{ref}_{I}(\mathbf{r}) = \sum_{i\in S_I}\vec{F}_i(\mathbf{r})\]r: coordinates of particles in an all-atom simulation
R: corresponding coarse-grained particle positions
The FM method finds \(\mathbf{F}^{CG}(\mathbf{R})\) that minimizes the objective function
\[\chi^2= \Bigg \langle \frac{1}{3N}\sum_{I=1}^{N}\left|\mathbf{F}^{CG}_{I}(\mathbf{R})-\mathbf{F}^{ref}_{I}(\mathbf{r})\right|^{2}\Bigg \rangle\]A 1 particle methanol system was used to learn the CG potential. The optimization was done at a learning rate of 0.1 using the ADAM optimizer37 by minimizing the objective function
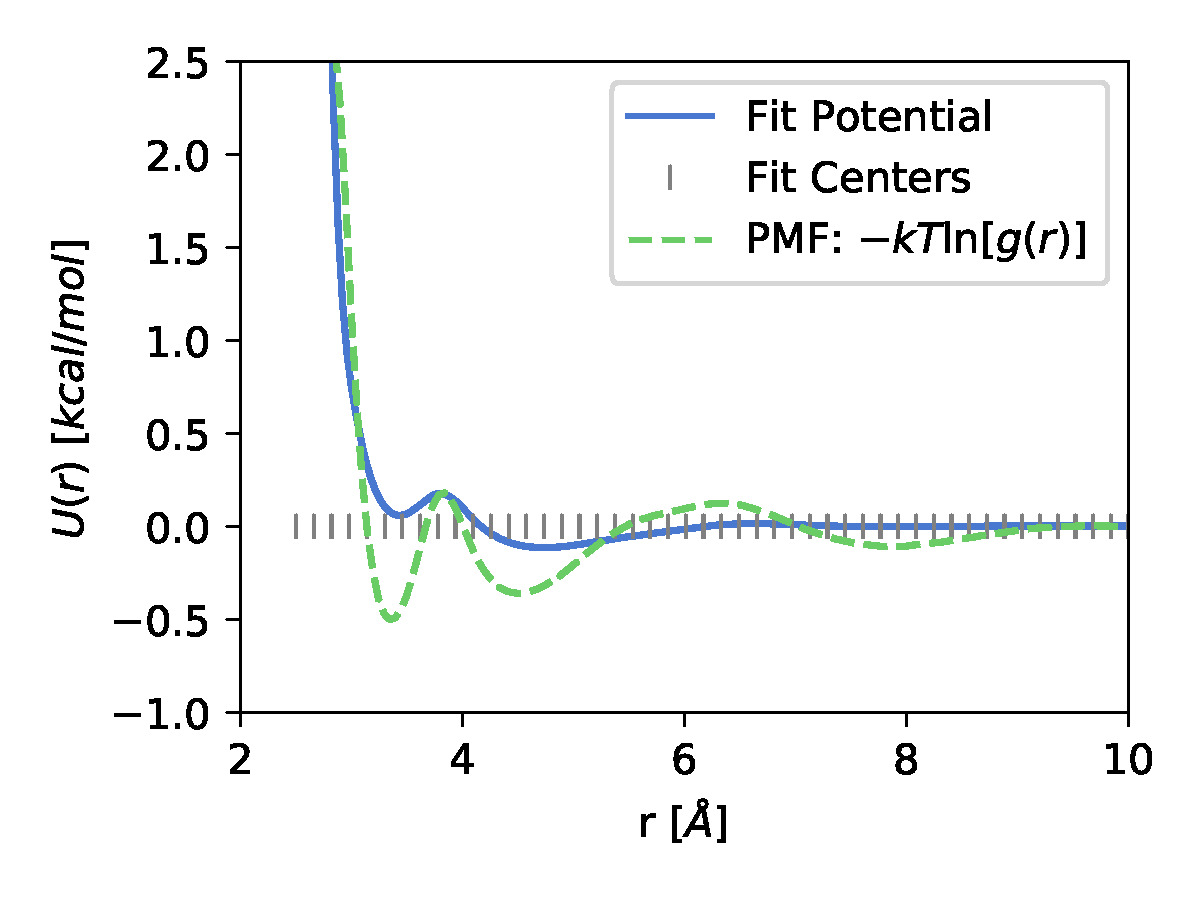 |
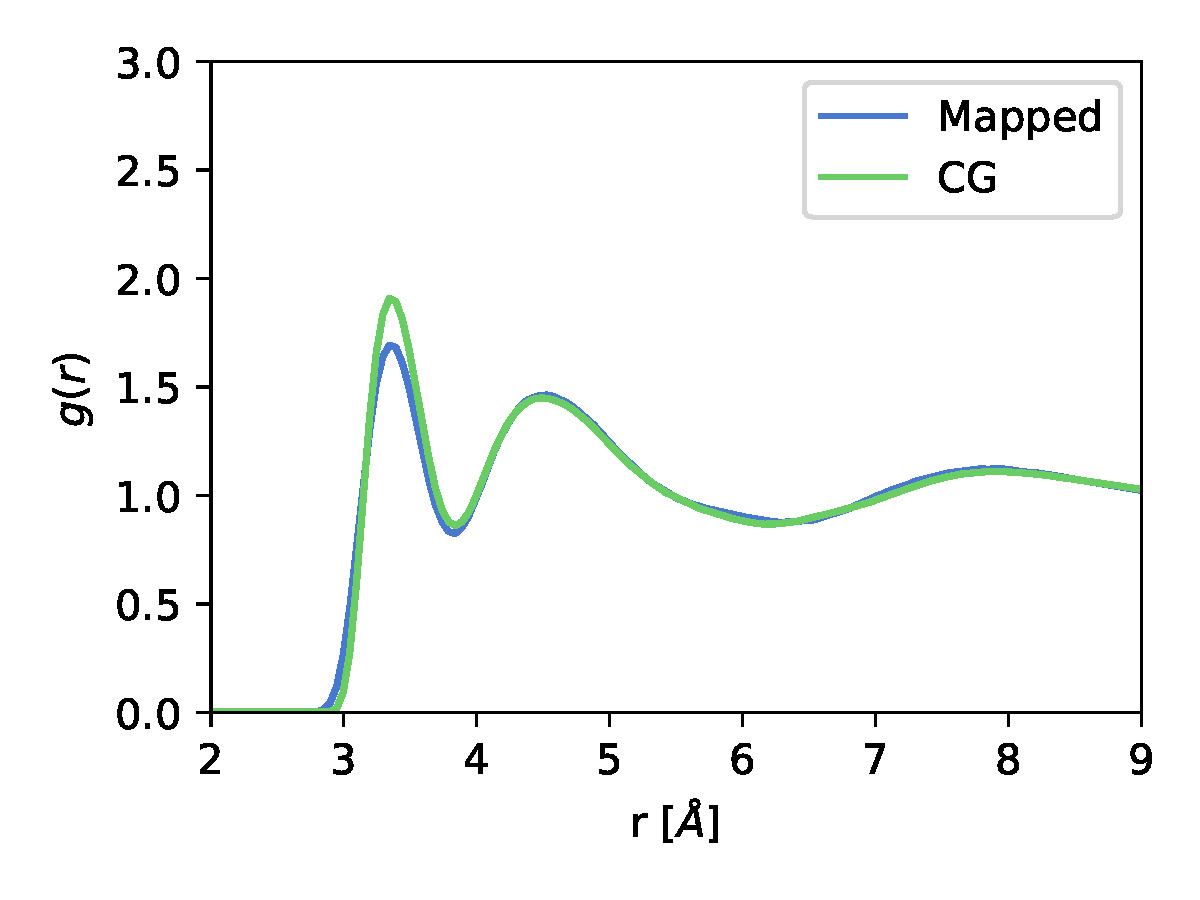 |
| The learned CG potential along with the fit centers of Guassian Basis set functions for a 1 particle methanol system | The center of mass radial distribution function from the mapped and CG trajectories. |
Source Code
Find the source code for HTF on github and the documentation is available here.
Future Work
We are working on expanding the suite of methods to perform CG potential learning implementations in HTF. Another goal for this project is to implement a neural network CG force-field to be able to get a function form of the potential rather than a tabulated form.
References
-
Barrett R, Chakraborty M, Amirkulova D, Gandhi HA, White AD (2019). A GPU-Accelerated Machine Learning Framework for Molecular Simulation: Hoomd-Blue with TensorFlow. ChemRxiv. doi: 10.26434/chemrxiv.8019527
-
Hadley KR and McCabe C (2012). Coarse-Grained Molecular Models of Water: A Review. Molecular simulation 38:671-681.
-
May A, Pool R, van Dijk E, Bijlard J, Abeln S, Heringa J, and Feenstra KA (2014). Coarse-grained versus atomistic simulations: realistic interaction free energies for real proteins. Bioinformatics 30:326-334.
-
Morriss-Andrews A and Shea JE (2014). Simulations of Protein Aggregation: Insights from Atomistic and Coarse-Grained Models. The Journal of Physical Chemistry Letters 5:1899{1908.
-
Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, Ghemawat S, Goodfellow I, Harp A, Irving G, Isard M, Jia Y, Jozefowicz R, Kaiser L, Kudlur M, Levenberg J, Mané D, Monga R, Moore S, Murray D, Olah C, Schuster M, Shlens J, Steiner B, Sutskever I, Talwar K, Tucker P, Vanhoucke V, Vasudevan V, Viégas F, Vinyals O, Warden P, Wattenberg M, Wicke M, Yu Y, and Zheng X (2015). TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems.
-
Anderson JA, Glaser J, and Glotzer SC (2019). HOOMD-blue: A Python package for high-performance molecular dynamics and hard particle Monte Carlo simulations. 10.1016/j.commatsci.2019.109363, arXiv:1308.5587.
-
Gowers RJ, Linke M, Barnoud J, Reddy TJE, Melo MN, Seyler SL, Dotson DL, Domanski J, Buchoux S, Kenney IM, Beckstein O (2016). MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In Benthall S and Rostrup S, editors, Proceedings of the 15th Python in Science Conference, 98-105, Austin, TX, 2016. SciPy, doi:10.25080/majora-629e541a-00e.
-
Michaud-Agrawal N, Denning EJ, Woolf TB, and Beckstein O (2011). MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32, 2319-2327, doi:10.1002/jcc.21787. PMCID:PMC3144279
Acknowledgements
Heta A. Gandhi was supported by a fellowship from The Molecular Sciences Software Institute under NSF grant OAC-1547580. The authors thank the Center for Integrated Research Computing (CIRC) at the University of Rochester for providing computational resources and technical support. This work was also supported by the National Science Foundation (CBET-1751471 and CHE-1764415).