
TCKDB - Theoretical Chemical Kinetics Database
Introduction
One of the grand visions in the chemical kinetics community is that kinetic models will be generated automatically (see Reaction Mechanism Generator [1]), while calculating important and sensitive thermodynamic properties and reaction rate coefficients on-the-fly using quantum chemical electronic structure methods.[2] The TCKDB project seamlessly integrates into this vision by making relevant high-quality data available, and mediating valuable information between different projects.
The goal of the TCKDB project is to establish a standardized open infrastructure for conveniently storing and attaining theoretical chemical information of relevance for kinetic computations. The chemical kinetics community is currently lacking such valuable tools.
The TCKDB project aims to (1) create a community-accepted standard for storing electronic structure calculations relevant to chemical kinetic modeling, and (2) allow sharing of and free access to such data for direct use in kinetic computations and modeling. Ultimately, the TCKDB will facilitate efficient workflows by negating the need for repeating the same computational tasks, giving access to fully detailed computations. As a by-product, it will facilitate big-data queries for either direct research or algorithm training purposes (e.g., learning conformational and transition state geometries). Finally, it would also serve as a citable repository that could become the standard when publishing kinetic-relevant electronic structure computations.
The big picture
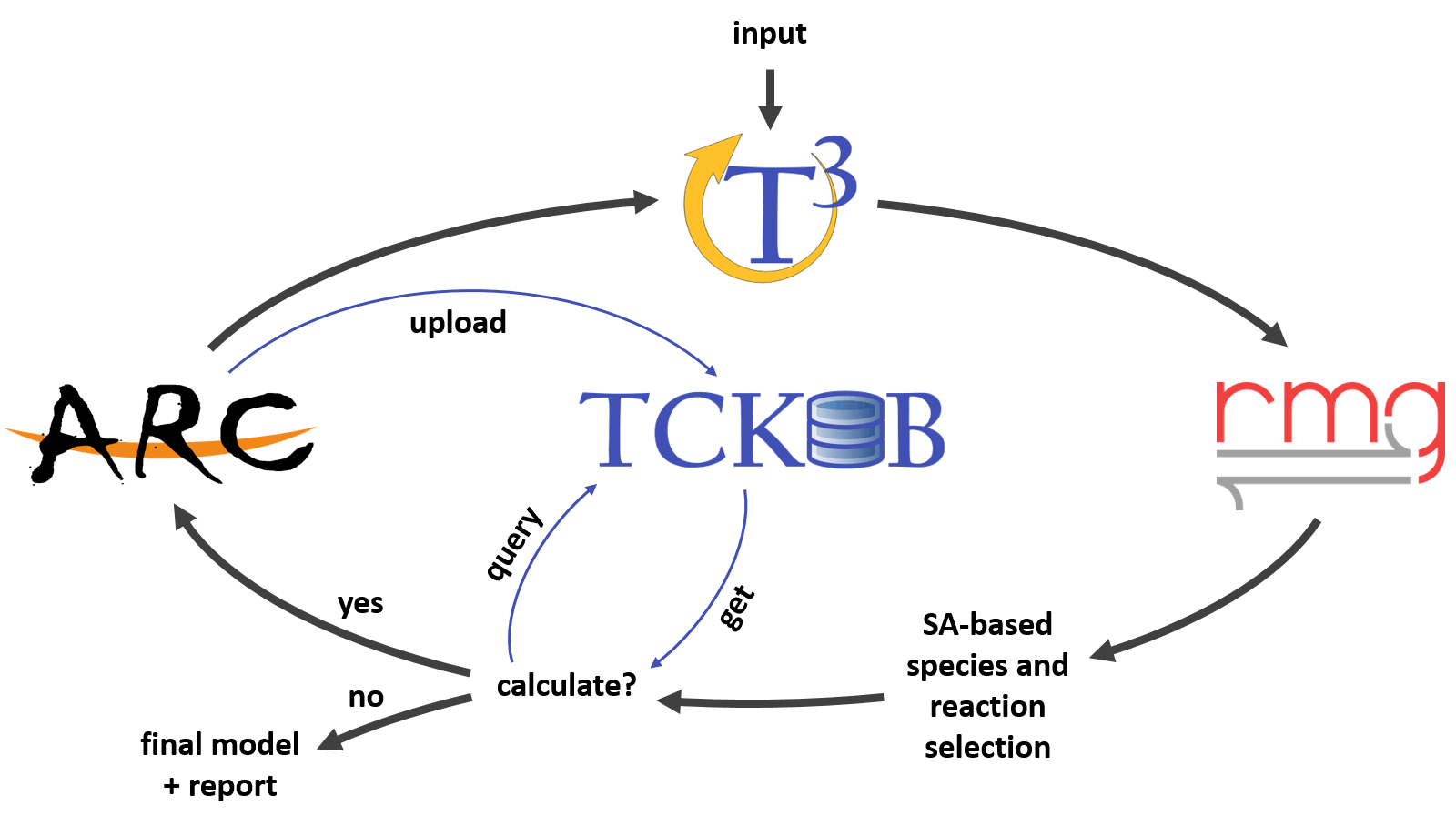
Figure 1: Envisioned automated kinetic model generation and refinement scheme.
The TCKDB project is intended to seamlessly integrate into an automated kinetic model generation and refinement scheme (Figure 1). Here, RMG (Reaction Mechnaism Generation) is a software for automatically generating chemical kinetic models, ARC (Automated Rate Calculator) is a software for automating electronic structure calculations and retrieve thermokinetic parameters, and T3 (The Tandem Tool) is a high-level routine that manages the iterative model generation and refinement operation. Before a calculation is requested, TCKDB will first be queried to check for data existence. If acceptable data exists, it will be used instead of consuming valueable computational resources for a reductant task. After a calculation is made, it is uploaded to TCKDB where it will undergo a review prcess and be integrated into the database for the benefit of the community.
Collaboration
Researchers across the field have already expressed interest in this project. Among them are Dr. Stephen J. Klippenstein (ANL), Dr. Judit Zádor (SNL), Prof. Carlo A. Cavallotti (Politecnico di Milano), Prof. Richard H. West (Northeastern U.), Dr. Brian Barnes (ARL), and Prof. William H. Green (MIT). The multi-PI interest in this project is expected to result in an immediate wide userbase via the different software being developed by these teams (KinBot,[3] EStokTP,[4] PACChem moldrive,[5] PAPR MESS,[6] AutoTST,[7] QTC,[2] RMG,[1] Arkane,[8] and ARC [9]).
Database structure
The primary tables in the database will be:
- Species
- Non physical species
- Reaction
- Pressure-dependent networks
Secondary tables include:
- Author
- Bot
- Literature
- energy transfer model
- L-J (Lannard-Jones coefficients)
- frequency scaling factor
- energy corrections
- levels of theory
- electronic structure software
Schema
A schema with moleculer properties for a Species object was created and will be considered to serve as a community standard. Following is an example of representing a Species in TCKDB:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
Species(label='formaldehyde',
statmech_software='Arkane (RMG) v3.0.0',
smiles='C=O',
charge=0,
multiplicity=1,
electronic_state='X',
coordinates={'symbols': ('C', 'O', 'H', 'H'),
'isotopes': (12, 16, 1, 1),
'coords': ((-0.0122240982, 0.0001804054, -0.00162116),
(1.2016481968, -0.0177341701, 0.1593624097),
(-0.5971643978, 0.9327281670, 0.0424401022),
(-0.5922597008, -0.9151744023, -0.2001813507))},
external_symmetry=2,
point_group='C2v',
conformation_method='ARC',
is_well=True,
is_global_min=True,
is_ts=False,
electronic_energy=-325.6458956547,
E0=123.54842,
frequencies=[132.2, 548.5, 1032.5, 2015.22, 2018.12, 3005.22],
scaled_projected_frequencies=[130.217, 540.2725, 1017.013,
1984.992, 1987.848, 2960.142],
normal_displacement_modes=[[0.125, 0.89, 0.35],
[-0.25, 0.25, -0.89],
[0.56, 0.98, -0.65],
[0.022, -0.005, 0.5],
[-0.98, -0.002, 0.65],
[0.05, 0.0025, -0.722]],
freq_id=11,
rigid_rotor='asymmetric top',
statmech_treatment='RRHO',
rotational_constants=[1.25, 8.56, 9.55],
H298=-109.4534,
S298=218.237,
Cp_values=[35.313, 39.037, 43.4299, 47.7813, 55.438, 61.463, 70.71],
Cp_T_list=[300, 400, 500, 600, 800, 1000, 1500],
heat_capacity_model={'model': 'NASA',
'T min': 100,
'T max': 5000,
'coefficients': {'low': [4.13878818E+00, -4.69514383E-03,
2.25730249E-05, -2.09849937E-08,
6.36123283E-12, -1.43493283E+04,
3.23827482E+00],
'high': [2.36095410E+00, 7.66804276E-03,
-3.19770442E-06, 6.04724833E-10,
-4.27517878E-14, -1.42794809E+04,
1.04457152E+01],
'T int': 1041.96}},
en_corr_id=33,
opt_path='path_opt',
freq_path='path_freq',
sp_path='path_sp')
Summary
It is our hope that once TCKDB is operable, it will be adopted by the kinetics community as a repository to store ab-initio thermokinetic parameters, and will assist in automation of chemical kinetic model generation.
References
- (a) C.W. Gao, J.W. Allen, W.H. Green, R.H. West, Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms, Computer Physics Communications 2016, 203, 221-225, doi: 10.1016/j.cpc.2016.02.013. (b) https://rmg.mit.edu/.
- M. Keçeli, S.N. Elliott, Y-P. Li., M.S. Johnson, C. Cavallottie, Y. Georgievskii, W.H. Green, M. Pelucchi, J.M. Wozniak, A.W. Jasper, S.J. Klippenstein, Automated computational thermochemistry for butane oxidation: A prelude to predictive automated combustion kinetics, Proceedings of the Combustion Institute 2019, 37, 363-371, doi: 10.1016/j.proci.2018.07.113
- Ruben Van de Vijver, Judit Zádor, KinBot: Automated stationary point search on potential energy surfaces, Computer Physics Communications, 248, 2020, https://doi.org/10.1016/j.cpc.2019.106947.
- C. Cavallotti, M. Pelucchi, Y. Georgievskii, S.J. Klippenstein, EStokTP: Electronic Structure to Temperature- and Pressure-Dependent Rate Constants—A Code for Automatically Predicting the Thermal Kinetics of Reactions, J. Chem. Theory Comput 2019, 15, 1122-1145, doi: 10.1021/acs.jctc.8b00701.
- https://github.com/PACChem/moldriver
- https://tcg.cse.anl.gov/papr/codes/mess.html
- P.L. Bhoorasingh, B.L. Slakman, F.S. Khanshan, J.Y. Cain, R.H. West, Automated Transition State Theory Calculations for High-Throughput Kinetics, J. Phys. Chem. A 2017, 121, 6896-6904, doi: 10.1021/acs.jpca.7b07361
- A. Grinberg Dana, M.S. Johnson, J.W. Allen, S. Sharma, S. Raman, M. Liu, C.W. Gao, C.A. Grambow, M.J. Goldman, D.S. Ranasinghe, R.J. Gillis, A.M. Payne, Y-P. Li, E.E. Dames, Z.J. Buras, N.M. Vandewiele, N.W. Yee, S.S. Merchant, B. Buesser, C.A. Class, C.F. Goldsmith, R.H. West, W.H. Green, Automated Reaction Kinetics and Network Exploration (Arkane): A Statistical Mechanics, Thermodynamics, Transition State Theory, and Master Equation Software, In preparation.
- A. Grinberg Dana, D., H. Wu, C. Grambow, X. Dong, M. Johnson, M. Goldman, M. Liu, W.H. Green, ARC - Automated Rate Calculator, version 1.1.0, https://github.com/ReactionMechanismGenerator/ARC, doi: 10.5281/zenodo.3356849
Acknowledgements
Alon Grinberg Dana was supported by a fellowship from The Molecular Sciences Software Institute under NSF grant OAC-1547580.