Extending Geometry Analysis
Overview
Teaching: 40 min
Exercises: 20 minQuestions
Write a function for calculating the angle between two points
Write a function which finds all bonds in a molecule.
Use values from a known source (QCArchive) to verify that function works correctly
For this lesson, we will extend the Geometry Analysis project which you completed as part of the pre-summer school workshop. We will write functions to calculate and store bonds in a molecule, and angles based on those bonds. Results will be verified using a known data set from QCArchive.
Using QCArchive
QCArchive is a software ecosystem from MolSSI for aggregating and sharing quantum chemistry data. We are going to pull some data from QCArchive today and use the functions we’ve used as analysis.
If you don’t have QCArchive installed, install it in your terminal using the command
$ conda install qcportal -c conda-forge
Once qcportal is installed, you can import the libraries for QCArchive. We will be using qcportal to pull data from the MolSSI instance of QCArchive.
# imports
import qcportal as ptl
# connect to qc Fractal
client = ptl.FractalClient()
FractalClient
Server: The MolSSI QCArchive Server
Address: https://api.qcarchive.molssi.org:443/
Username: None
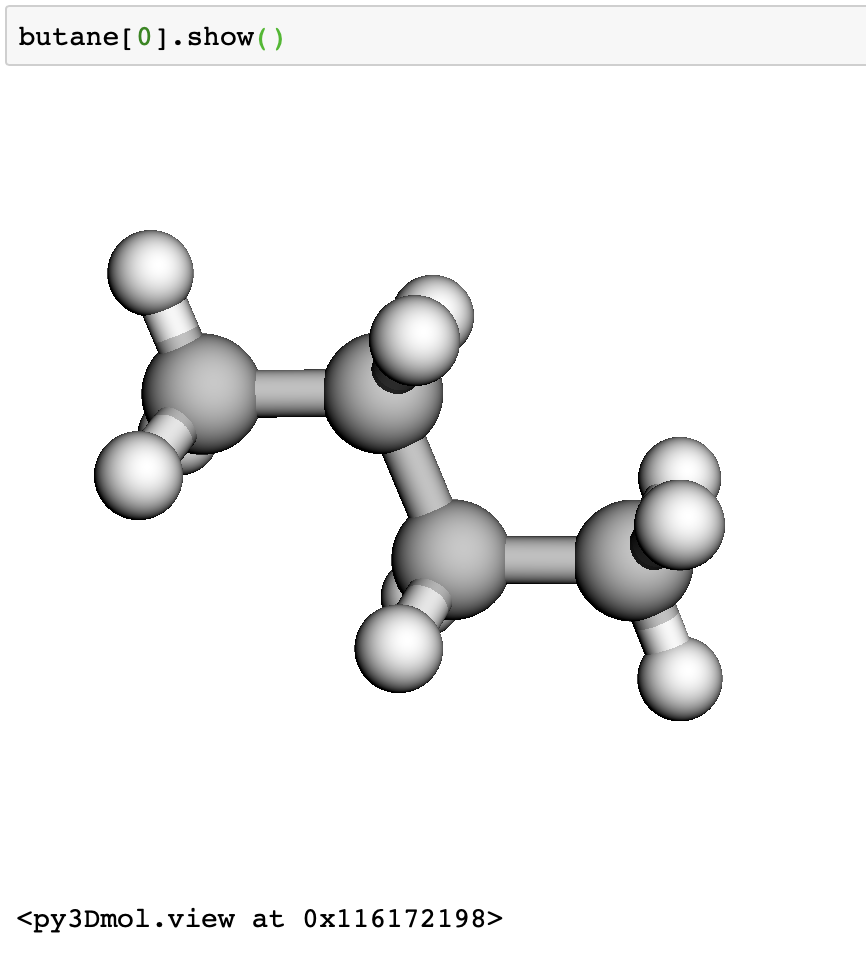
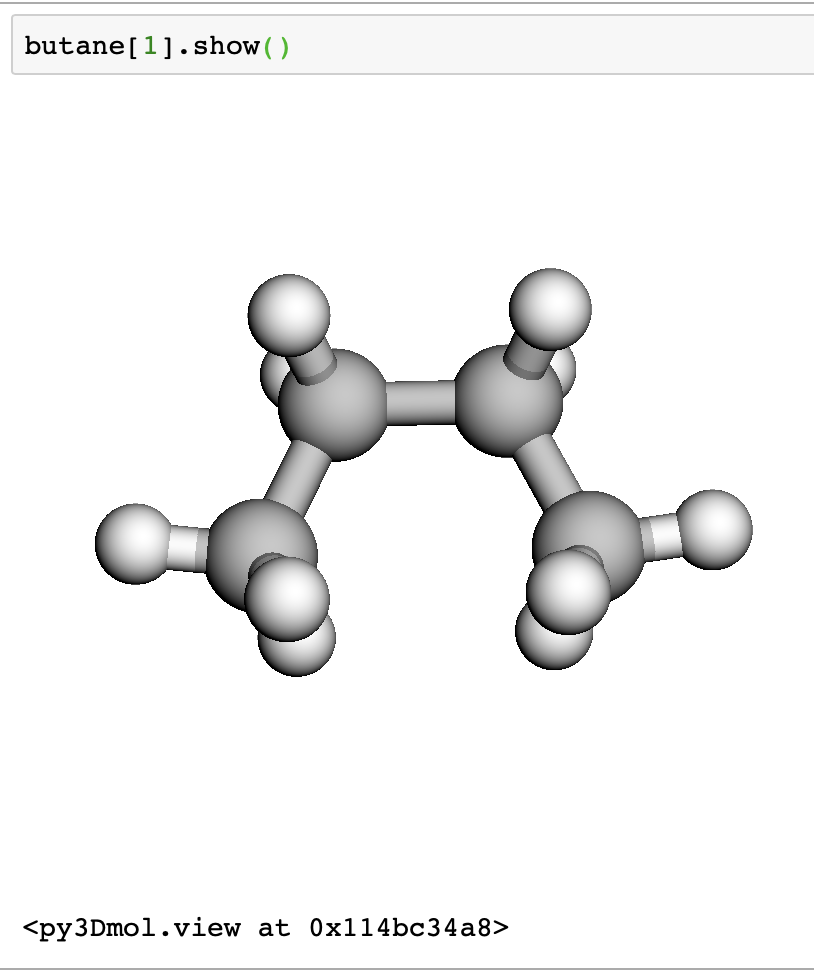
Next, we grab some butane molecules using their ids. We are getting a trans and cis butane, ids 61139 and 70659.
butane = client.query_molecules(id=['61139', '70659'])
This has given us a list of QCArchive molecules.
butane
[<Molecule(name='C4H10' formula='C4H10' hash='3bbc6db')>,
<Molecule(name='C4H10' formula='C4H10' hash='bb665a3')>]
We can view the structures interactively in a jupyter notebook.
butane[0].show()
butane[1].show()
Let’s look at some of the methods and attributes associated with each molecule.
dir(butane[0])
['Config',
'__abstractmethods__',
'__annotations__',
'__class__',
'__config__',
'__delattr__',
'__dict__',
'__dir__',
'__doc__',
'__eq__',
'__fields__',
'__fields_set__',
'__format__',
'__ge__',
'__get_validators__',
'__getattr__',
'__getattribute__',
'__getstate__',
'__gt__',
'__hash__',
'__init__',
'__init_subclass__',
'__iter__',
'__le__',
'__lt__',
'__module__',
'__ne__',
'__new__',
'__reduce__',
'__reduce_ex__',
'__repr__',
'__setattr__',
'__setstate__',
'__sizeof__',
'__slots__',
'__str__',
'__subclasshook__',
'__validators__',
'__values__',
'__weakref__',
'_abc_impl',
'_calculate_keys',
'_custom_root_type',
'_decompose_class',
'_get_key_factory',
'_get_value',
'_inertial_tensor',
'_iter',
'_json_encoder',
'_orient_molecule_internal',
'_repr_html_',
'_schema_cache',
'align',
'atom_labels',
'atomic_numbers',
'comment',
'compare',
'connectivity',
'construct',
'copy',
'dict',
'extras',
'fields',
'fix_com',
'fix_orientation',
'fix_symmetry',
'fragment_charges',
'fragment_multiplicities',
'fragments',
'from_data',
'from_file',
'from_orm',
'geometry',
'get_fragment',
'get_hash',
'get_molecular_formula',
'hash_fields',
'id',
'identifiers',
'json',
'json_dict',
'mass_numbers',
'masses',
'measure',
'min_zero',
'molecular_charge',
'molecular_multiplicity',
'must_be_3n',
'must_be_n',
'must_be_n_frag',
'name',
'nelectrons',
'nuclear_repulsion_energy',
'orient_molecule',
'parse_file',
'parse_obj',
'parse_raw',
'populate_real',
'pretty_print',
'provenance',
'real',
'schema',
'schema_json',
'schema_name',
'schema_version',
'scramble',
'show',
'symbols',
'to_file',
'to_string',
'update_forward_refs',
'validate']
Try out some of these, what interesting features can you find?
For our exercises today, we will be using the atomic coordinates. These are stored in the geometry attribute as a NumPy array.
Let’s grab both of these in a list
butane_geometries = [butane[0].geometry.copy(), butane[1].geometry.copy()]
Returning to geometry analysis
Recall from the previous lesson that we have a function for calculating distances
def calculate_distance(rA, rB):
"""Calculate the distance between points A and B"""
dist_vec = (rA-rB)
distance = np.linalg.norm(dist_vec)
return distance
In the geometry analysis project, you also had a script which looped over points in a molecule to find the bonds. Let’s define this function.
def build_bond_list(coordinates, max_bond=1.55, min_bond=0):
num_atoms = len(coordinates)
for atom1 in range(num_atoms):
for atom2 in range(atom1, num_atoms):
distance = calculate_distance(coordinates[atom1], coordinates[atom2])
if distance > min_bond and distance < max_bond:
print(F'{atom1}, {atom2} : distance')
Exercise
Modify the
build_bond_listfunctions so that the function returns a dictionary which has keys of the atom indices, and values of bond lengths.The keys should be something which you can use to index back into the coordinates array.
Answer
def build_bond_list(coordinates, max_bond=1.55, min_bond=0): num_atoms = len(coordinates) bonds = {} for atom1 in range(num_atoms): for atom2 in range(atom1, num_atoms): distance = calculate_distance(coordinates[atom1], coordinates[atom2]) if distance > min_bond and distance < max_bond: bonds[(atom1, atom2)] = distance return bonds
Python Dictionary Keys
To make the solution store the atom indices as keys, we used a tuple. What happens if you try using a list?
Try using a list as keys (ie
[atom1, atom2]) as keys.If you do this, you get a
TypeError. This is because dictionary keys must be immutable types. Thus, you can use tuples, strings, or numeric types as keys, but you can’t use lists.
Let’s try this on our butane geometry we pulled from QCArchive.
bond_list = build_bond_list(butane_geometries[0])
print(bond_list)
{}
We aren’t getting any bonds - does this seem right?
Let’s troubleshoot by querying the butane molecules from QCArchive.
butane[0].symbols
['C', 'C', 'C', 'C', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H']
Even though we’ve written our own distance calculation, QCArchive actually has one built in. Let’s look at it’s value for the first two Carbon atoms.
qcarchive_distance = butane[0].measure([0, 1])
print(qcarchive_distance)
7.31415824856433
my_distance = calculate_distance(butane_geometries[0][0], butane_coordinates[0][1])
7.314158248564329
This is a good sign - we are getting the same as answer as QCArchive expects.
The default bond distance value of 1.55 is appropriate if your units are angstrom. However, QCArchive stores distances in units of bohr.
We can do this conversion ourselves by googling a conversion factor, but we will another utility associated with QCArchive called qcelemental to get the exact conversion factor.
import qcelemental
angstrom_to_bohr = qcelemental.constants.conversion_factor("angstrom", "bohr")
print(angstrom_to_bohr)
max_length = 1.55 * angstrom_to_bohr
butane_bond = build_bond_list(butane_geometries[0], max_bond=max_length)
In the next cell, look at butane_bonds
butane_bonds
{(0, 2): 2.873020656801253,
(0, 4): 2.068839651551603,
(0, 5): 2.068484041315794,
(0, 6): 2.068821759435787,
(1, 3): 2.873059651780396,
(1, 7): 2.068247482474781,
(1, 8): 2.0685780442929644,
(1, 9): 2.0687346633381822,
(2, 3): 2.8862132661474598,
(2, 10): 2.0715801221019885,
(2, 11): 2.0716218178677623,
(3, 12): 2.0713472807014583,
(3, 13): 2.071754810836534}
Let’s compare this to the connectivity QCArchive has stored.
butane[0].connectivity
[(0, 2, 1.0),
(0, 4, 1.0),
(0, 5, 1.0),
(0, 6, 1.0),
(1, 3, 1.0),
(1, 7, 1.0),
(1, 8, 1.0),
(1, 9, 1.0),
(2, 3, 1.0),
(2, 10, 1.0),
(2, 11, 1.0),
(3, 12, 1.0),
(3, 13, 1.0)]
The first two elements of each tuple are the atom indices, and the third is a bond order
## Applying our functions to a different molecule
Next, we will look at a different kind of record in QCArchive. We are going to examine a geometry optimization.
opt = client.query_procedures(id=2658710)[0]
We can examine this object
dir(opt)
We can view the initial and final molecules
initial = opt.get_initial_molecule()
initial
final = opt.get_final_molecule()
final
We can access the coordinates in the same was as before.
initial.geometry
However, you will notice that no connectivity is defined for these molecules.
initial.connectivity
initial_bonds = build_bond_list(initial.geometry, max_bond=max_length)
final_bonds = build_bond_list(final.geometry, max_bond=max_length)
Exercise
Write a loop which will print the change in bond length from the initial configuration to the final configuration.
Answer
for k in initial_bonds: diff = final_bonds[k] - initial_bonds[k] print(k, diff)
Now that we have a function for calculating distance, let’s also create a function for calculating angles. We know that we can calculate the angle between two vectors using the dot product.
Exercise
Write a function called
calculate_anglewhich takes three points (numpy arrays) as input, and returns the angle measurement between the points. Next, add an optional argument to the functiondegreeswhich will allow the user to return the angle in degrees.Hint
You will use the numpy functions
np.arccos,np.dot,np.linalg.norm, andnp.degrees.Answer
def calculate_angle(rA, rB, rC, degrees=False): """Calculate angle between points A, B, and C""" AB = rB - rA BC = rB - rC theta = np.arccos(np.dot(AB, BC) / (np.linalg.norm(AB)*np.linalg.norm(BC))) if degrees: return np.degrees(theta) else: return theta
Key Points